
Журнал «Внутренняя медицина» 4(4) 2007
Вернуться к номеру
Антитромбоцитарне лікування хворих, у яких на фоні приймання аспірину розвинувся гострий коронарний синдром з елевацією сегмента ST, асоційований з артеріальною гіпертензією
Авторы: Т.Й. Мальчевська, к.м.н., Г.І. Мишанич, Національний медичний університет імені О.О. Богомольця, кафедра пропедевтики внутрішніх хвороб № 1
Рубрики: Медицина неотложных состояний, Кардиология
Разделы: Справочник специалиста
Версия для печати
Артеріальна гіпертензія (АГ) є незалежним фактором ризику серцево-судинних ускладнень як у популяції взагалі, так і серед постінфарктних пацієнтів зокрема. Існують докази того, що за наявності АГ у постінфарктних пацієнтів частота повторних інфарктів міокарда (ІМ), серцево-судинна і загальна летальність є суттєво вищими порівняно з пацієнтами із нормальним рівнем артеріального тиску (АТ). Пацієнти, які перенесли гострий ІМ (ГІМ), є групою високого ризику повторних інцидентів. Постійне ретельне приймання антигіпертензивних препаратів є невід'ємною стратегією ведення постінфарктних пацієнтів із АГ. У лікуванні постінфарктних хворих із АГ препаратами вибору є ІАПФ, β-адреноблокатори, до альтернативних відносять АРАІІ. Обов'язковим є призначення й гіполіпідемічної терапії. На сьогодні антитромбоцитарні агенти стали обов'язковим компонентом не лише первинної, а й вторинної профілактики атеросклеротичних захворювань, причому їх призначення за артеріальної гіпертензії вимагає контролю її перебігу. Ера аспірину як антитромбоцитарного препарату розпочалася з 1949 р., коли його призначення як протизапального засобу вірогідно знижувало смертність від інфаркту міокарда у чоловіків [1]. Ефективність АСК доведена численними клінічними дослідженнями. За даними метааналізу 287 досліджень, проведеного групою експертів Antiplatelet Trialists Collaboration (2002), тривале приймання АСК (75–325 мг на добу) знижує частоту нефатального інфаркту міокарда на 1/3, нефатального інсульту — на 1/4 і судинної смертності — на 1/6, тобто сумарна частота серйозних судинних подій зменшується на 25 % [2]. Ці дані лягли в основу широкого впровадження АСК (аспірин) у стандартні режими вторинної профілактики серцево-судинних ускладень.
Щодо механізмів біотрансформації циклооксигенази під впливом АСК зазначимо, що циклооксигеназа-1 (ЦОГ-1, простагландинсинтаза) каталізує трансформацію арахідонової кислоти (АА) в нестабільну проміжну форму PGH2 [3]. Надалі тромбоксансинтаза впливає на PGH2 до утворення тромбоксану (ТХА2), що індукує агрегацію тромбоцитів і є сильним вазоконстриктором. Аспірин втручається переважно в біосинтез циклічних простаноїдів: ТХА2, простацикліну та інших простагландинів. Він необоротно інгібує ЦОГ-1 ацетилюванням серину в положенні 530 й індукує тривалий функціональний дефект тромбоцитів. Результуюче зниження продукції простагландинів і ТХА2, можливо, значною мірою вважається аспіриновим антитромбоцитарним ефектом [1, 2]. З огляду на те, що ТХА2 і PGI2 мають протилежні гемостатичні ефекти, дані підтверджують, що антитромбоцитарний ефект тромбоксанової інгібіції домінує над можливим протромботичним ефектом інгібіції PGI2 [2]. Діапазон застосованих антитромботичних доз аспірину становить у середньому 80–500 мг на добу. Проте при тривалому застосуванні низьких доз аспірину спостерігається кумулятивна природа інгібіції ТХА2 генерації тромбоцитів, оскільки інгібіція ЦОГ-1 АСК є необоротною [5]. Відсутній прямий зв'язок між генерацією тромбоцитами ТХА2 та інгібіцією тромбоксанзалежної агрегації тромбоцитів. Функціональна здатність тромбоцитів порушується за інгібіції більше ніж 95 % ТХА2-генерації [6]. Рівень тромбоксану тромбоцитів падає нижче 95 % тільки після декількох днів застосування аспірину в низьких дозах. Проте низькі дози (40–60 мг), як і високі (500 мг), пригнічують тромбоксановий синтез більше ніж на 95 % [7, 8]. Хоча аспірину у високих дозах властива протизапальна дія внаслідок інгібіції ЦОГ-2, все ж таки інгібіція ЦОГ-1 у 170 разів більш виражена порівняно з інгібіцією ЦОГ-2 [9]. Низькі дози АСК із незначним протизапальним ефектом залишають простациклінову судинну формацію майже інтактною [10]. Дози аспірину вищі за 80 мг/добу частково інгібують ендогенний простацикліновий біосинтез [11]. Було продемонстровано 81–100% інгібіцію простациклінового синтезу через 2 години після всмоктування 150 мг АСК [1].
Епідеміологічні дослідження показали, що як дуже низькі, так і високі дози АСК (30–1500 мг) еквівалентні щодо антитромбоцитарного ефекту й інгібіція тромбоцитарної ЦОГ-1 є дійсно остаточною метою дії аспірину [10]. Це підтверджує точку зору, що інгібіція ЦОГ-1 тромбоцитів низькими дозами АСК є достатньою для пояснення кардіопротективної дії аспірину в проведених дослідженнях [1].
Тим не менше Syrbe вказує, що індивідуальна доза АСК видається необхідною для повної інгібіції агрегації тромбоцитів ex vivo. Так, встановлення дози АСК для пацієнтів із серцево-судинними захворюваннями (ССЗ) є необхідним для попередження недостатності терапії аспірином або так званої аспіринорезистентності (АР).
Аспірин, необоротно інгібуючи ЦОГ-1, впливає лише на тромбоксановий шлях активації тромбоцитів, тобто ТХА2 [20]. Проте тромбоцити можуть активуватися іншим шляхом, що не блокується аспірином, і таким шляхом є АДФ. Дослідники передбачають, що тромбоцити в аспіринорезистентних пацієнтів проявляють гіперчутливість до АДФ, що може пояснювати аспіринорезистентність і терапевтичне покращання через альтернативне застосування різних антитромбоцитарних агентів. В іншому дослідженні пацієнти з підозрою на ішемічну хворобу серця (ІХС) класифікувалися відповідно до числа стенозованих судин, індукованої агрегації тромбоцитів. Результати свідчать, що аспірин недостатньо інгібує АДФ і колагеніндуковану агрегацію тромбоцитів у пацієнтів із ССЗ. Результати підтвердили точку зору про те, що нетромбоцитарноопосередковані ефекти АСК можуть бути клінічно більш важливими, ніж антитромбоцитарний ефект [28]. І зовсім недавно, окрім протизапального й антитромбоцитарного впливу, встановлено покращання ендотеліальної функції аспірином, а при монотерапії він вірогідно знижує як систолічний, так і діастолічний АТ. Причому встановлена часозалежність такої дії, що вірогідно проявлялася гіпотензивним ефектом під час застосування його у вечірні години, а не після пробудження.
З огляду на останні рекомендації Європейського кардіологічного товариства та Американського коледжу кардіологів щодо ведення пацієнтів із гострим коронарним синдромом (ГКС) без елевації сегмента ST як антитромбоцитарні засоби для тривалого застосування визнані як аспірин, так і похідний тієнопіридинів — клопідогрель та їх комбінація. Більше того, в останніх рекомендаціях із ведення пацієнтів із стабільною стенокардією на першому місці лікувальної стратегії теж перебувають антитромбоцитарні препарати. Аспірин став еталоном у лікуванні ішемічних синдромів. Але за деяких умов пацієнти вживати аспірин не можуть (алергія, аспіринова астма, НПЗП-індукована гастропатія, виразкова хвороба, шлунково-кишкова кровотеча); встановлено, що АСК не завжди захищає пацієнтів від серцево-судинних катастроф [9, 11]. Ось чому в науковій літературі з'явилося поняття аспіринорезистентності, точніше феномену медикаментозної недостатності аспірину [4, 5, 7, 9]. Аспіринорезистентність асоціюється з підвищеним ризиком виникнення раптової серцевої смерті, ІМ та цереброваскулярних подій [10]. За даними літератури, у 5–40 % пацієнтів спостерігається відсутність антитромбоцитарного ефекту аспірину, проте залежно від коагулологічних методів інтерпретації аспіринорезистентності ця цифра може коливатися. Під час використання неспецифічних тестів тромбоцитарної активації/агрегації з моніторингу дії аспірину, за даними Campbell і Steinhubl, що спираються на результати 11 досліджень, інциденти аспіринорезистентності варіюють від 5,5 до 61 % [25, 26]. Дослідження, що спиралися на визначення TХB2, аспіринорезистентність виявляли в 1–1,7 % пацієнтів, і в дослідженнях, де оцінювалась арахідоніндукована агрегація тромбоцитів, аспіринорезистентність складала менше 1 % [25, 26]. Отже, існує потреба не тільки застосування альтернативного препарату у випадках, коли аспірин протипоказаний, але й посилення його ефекту на тромбоцити для досягнення більш повного захисту від внутрішньосудинних тромбозів. Останнім часом широкого застосування набули препарати групи тієнопіридинів, зокрема клопідогрель.
Клопідогрель належить до тієнопіридинів, в основі його дії лежить антагонізм до аденозин-5-дифосфат рецепторів. Він є необоротнім інгібітором тромбоцитів і діє впродовж усього періоду їх життя. Клопідогрель необоротно з'єднується з рецептором P2Y12, цей підтип АДФ-рецепторів пов'язаний із посиленням агрегації і секреції. Блокуючи рецептори до АДФ, препарати цієї групи запобігають внутрішньоклітинному виникненню сигналів, що врешті-решт призводить до активації GP IIb/IIIa рецепторів тромбоцитів. Крім цього, запобігаючи цим сигналам тієнопіридини гальмують і експресію адгезивних молекул на поверхні тромбоцитів.
Тієнопіридини не втручаються в обмін арахідонової кислоти, а отже, не можуть втручатися в обмін простацикліну судинної стінки. На останнє здатний аспірин, і це теоретично вважається його недоліком. Гальмуючи ефекти АДФ, що вивільняється з гранул, і перешкоджаючи подальшому вивільненню вмісту цих гранул, тієнопіридини перешкоджають агрегації, що викликається й іншими агоністами, такими як фактор активації тромбоцитів, колаген, низькі концентрації тромбіну.
У печінці під впливом ферментної системи цитохром Р450-3А утворюються декілька активних метаболітів клопідогрелю. На АДФ-рецептори діє не сам клопідогрель, а один із його метаболітів. Основному метаболіту клопідогрелю, що доступний визначенню у фармакокінетичних дослідженнях, антитромбоцитарна дія не властива. Антитромбоцитарний ефект обумовлюють короткоживучi активнi метаболiти, і він настає повiльнiше. Великомасштабними дослідженнями було доведено, що для досягнення швидкого ефекту необхiднi ударнi дози, зокрема для клопідогрелю — 300 мг [6]. Було продемонстровано, що антитромбоцитарний ефект такої дози препарату на фоні вживання аспірину в людини проявляється вже через 90 хв, а через 6 год сягає такого ж рівня, як після 10-денного тривалого одночасного приймання клопідогрелю й аспірину. Після приймання per os 75 мг клопідогрелю він швидко всмоктується, і пік концентрації в плазмі його основного, але неактивного метаболіту спостерігається вже через 1 год, а гальмування функції тромбоцитів відбувається значно пізніше.
Щодо застосування клопідогрелю в особливої категорії пацієнтів, то слід відзначити, що в людей віком понад 75 років концентрація в плазмі крові метаболіту клопідогрелю значно вища, ніж у молодих, але це не супроводжується відмінностями в агрегації тромбоцитів і часі кровотечі і не вимагає зміни дозування залежно від віку [10]. У пацієнтів із суттєвим зниженням функції нирок (ХНН) рівень метаболіту клопідогрелю значно нижчий, ніж у пацієнтів із помірними порушеннями чи у здорових. Гальмування агрегації тромбоцитів у хворих із ХНН на 25 % менш виражене, ніж у здорових. У той же час залежності змін часу кровотечі від стану функції нирок не було виявлено, що й стало підставою для рекомендацій не змінювати дозу клопідогрелю у хворих з ураженням нирок. Досвід застосування клопідогрелю у хворих із вираженим порушенням функції печінки (з можливим геморагічним діатезом) практично відсутній. У пацієнтів, які мають виразкове ураження шлунково-кишкового тракту, за даними консенсусів із ведення пацієнтів, як за гострого коронарного синдрому, так і за стабільного перебігу ІХС клопідогрель у запобіганні атеротромботичних інцидентів визнаний альтернативою протипоказаному аспірину через високий ризик геморагічних ускладнень під час застосування останнього [6, 9, 13]. Щодо групи високого ризику (це пацієнти з цукровим діабетом), то їм показане профілактичне застосування не лише аспірину, а і його комбінації з клопідогрелем через синергізм антитромбоцитарного ефекту.
Ефективність клопідогрелю була доведена клінічними дослідженнями САРRIЕ, CREDO, PCI CURE, CURE, СLАSSIСS Сhinese cardiac study (CCS-2), COМMIT, CLARITY, WATCH, CHARISMA. У керівництвах із лікування ГКС без підйому ST рекомендовано застосовувати клопідогрель тоді, коли неможливо використовувати аспірин. Але більш визначеним стало місце клопідогрелю в лікуванні ГКС без підйому ST після закінчення дослідження СURE. Це дослiдження, що вивчало ефект клопiдогрелю при нестабiльнiй стенокардiї та не-Q-IМ (2001), виявило, що при ГКС терапія клопiдогрелем у поєднаннi з аспiрином є ефективнiшою, нiж монотерапiя аспiрином. У групi клопiдогрель + аспiрин порiвняно із групою, яка вживала тільки аспірин, на 20 % знижувався вiдносний ризик серцево-судинної смертi, IМ, iнсульту — 1-ї первинної точки (р < 0,001) та на 16 % — ризик серцево-судинної смертi, IМ, iнсульту та рефрактерної iшемiї — 2-ї первинної точки (р < 0,001).
Остаточно причини виникнення резистентності до антитромбоцитарних препаратів залишаються не з'ясованими. Хоча в останні роки доведений зв'язок між аспіринорезистентністю [19] і генетичною схильністю до поліморфізму тромбоцитів (алель PLA2 GP ІІІа). Не останню роль у цьому біохімічному феномені відіграє найбільша спорідненість тромбоцитів до фібриногену. Доведено, що серед пацієнтів із більшим рівнем фібриногену є вищим відсоток аспіринорезистентних. Передбачається, що аспіринорезистентність може бути спричинена збільшенням чутливості тромбоцитів до колагену і значно вищим рівнем Р-селектину [27]. Причини аспіринорезистентності криються також у низькому комплайєнсі пацієнтів, недостатній дозі аспірину, медикаментозній взаємодії з іншими препаратами, зокрема з нестероїдними протизапальними препаратами, що для пацієнтів із супутніми дифузними захворюваннями сполучної тканини є вкрай необхідними. Відомим є той факт, що кардіопротекція аспірину різко знижується при одночасному його застосуванні, зокрема, з ібупрофеном.
Останнім часом встановлений зв'язок АР із гіперхолестеринемією. Також збільшується відсоток аспіринорезистентних пацієнтів із підвищенням рівня фізичного навантаження. Нещодавно було встановлено, що серед пацієнтів, які були аспіринорезистентні чи напіврезистентні, переважають жінки і ті, хто менше палили, порівняно з аспіриночутливими. Щодо жінок, то вони були більш резистентними до аспірину порівняно з чоловіками: 67,7 і 26,6 % відповідно (р = 0,02), нижчим був у них і рівень гемоглобіну, можливо, цим пояснюються менша аспіринокардіопротекція в жінок і недостатній захист від імовірного виникнення першого інфаркту міокарда Вік є одним із домінуючих факторів ризику ішемічної хвороби серця, і смертність від інфаркту міокарда також збільшується з віком. Простежується тендеція залежності аспіринорезистентності й напіврезистентності від збільшення віку пацієнтів, тоді як у пацієнтів із різною расовою приналежністю, із захворюваннями нирок і печінки відсутня різниця в чутливості до приймання аспірину й кількості тромбоцитів. Частіше аспіринорезистентність зустрічалась у пацієнтів із цукровим діабетом 2-го типу. Отже, безліч факторів можуть ініціювати аспіринорезистентність.
Останнім часом встановлено, що половина пацієнтів, які є аспіринорезистентними, є резистентними також і до клопідогрелю [12, 13, 17]. Зокрема, серед пацієнтів із цукровим діабетом, які підлягали інтервенційним перкутанним втручанням, стентуванню, виявилося більше резистентних до клопідогрелю, у них відзначався більший індекс маси тіла. Ось чому для запобігання або уникнення резистентності до клопідогрелю в цієї категорії пацієнтів доза останнього має бути дещо вищою [15]. Клопідогрельрезистентність тісно корелює з інсулінорезистентністю і рівнем глікозильованого гемоглобіну. Несподіваними виявилися результати щодо факту, що паління підвищує ефективність клопідогрелю. Підвищення антитромбоцитарного ефекту клопідогрелю автори пояснюють конвертацією його в активну форму системою цитохром P450, що активується поліциклічним ароматичним гідрокарбонатом, який вивільнюється сигаретним димом. Клопідогрельрезистентність асоціюється з високим ризиком кардіоваскулярних подій, вірогідним відсотком пацієнтів, хворих на ІМ з елевацією сегмента ST (STEMI) [13, 17]. Хоча, на думку інших авторів, немає вірогідної відмінності між профілем відповіді тромбоцитів у пацієнтів як із STEMI, так і без STEMI. Труднощі полягають ще в тому, що не завжди ті багатогранні процеси, що відбуваються in vivo, можна відтворити in vitro, і виміри активності тромбоцитів традиційно є важкими в інтерпретації клінічних подій.
Останнім часом жваво обговорюється питання подвійної резистентності як до аспірину, так і до клопідогрелю. Так, половина пацієнтів, які були аспіринорезистентними, виявилися резистентними і до клопідогрелю, у цих пацієнтів відзначався і високий рівень МВ КФК.
Комбінація клопідогрелю й аспірину визнана стандартом у профілактиці підгострого тромбозу стенту й захисту від ішемічних подій у різних клінічних ситуаціях, включаючи гострий коронарний синдром і STEMI. Хоча досвід деяких авторів показує, що, незважаючи на агресивну терапію, деякі пацієнти не відповідають на неї і оборотність подій є звичним явищем. Так, дослідження SYNERGY (New Strategy of Enoxaparin Revascularisation and Glycoprotein IIb/IIIa Inhibitors) показало, що в пацієнтів із високим ризиком ГКС, незважаючи на агресивну потрійну антитромбоцитарну терапію аспірином, клопідогрелем та інгібіторами GР IIb/IIIa рецепторів, зустрічалися рецидиви ІМ упродовж 30 днів. Тому постало питання щодо потрійної резистентності, особливо до інгібітора GР IIb/IIIa рецепторів абсиксимабу.
Щодо комбінованого застосування, то впродовж останніх років активно обговорюється питання про можливість зниження сприятливих ефектів ІАПФ під впливом аспірину та при їх поєднаному застосуванні, а це частіше стосується пацієнтів з постінфарктним кардіосклерозом і супутніми артеріальною гіпертензією, серцевою недостатністю, для яких ця група препаратів є препаратами вибору. Одним із компонентів антигіпертензивного й вазопротективного впливу ІАПФ є збільшення синтезу вазодилататорних простагландинів, таких як PGЕ2. У той же час аспірин інгібує дозозалежне впродовж 4–6 годин інгібування простагландинів (у тому числі PGЕ2), що є складовою його протизапального ефекту. Деякими авторами відзначено, що комбіноване застосування АСК та ІАПФ, можливо, знижує позитивний вплив останніх на прогноз у хворих із серцевою недостатністю і в постінфарктних хворих.
При цьому для досягнення повноцінної антитромбоцитарної відповіді достатніми дозами аспірину вважаються 80–100 мг на добу, які є досить низькими для суттєвого впливу на синтез PGЕ2. Для запобігання можливому антагонізму між препаратами слід зберігати часові проміжки в прийманні АСК та ІАПФ від 6 до 12 годин.
Суперечливим видалися взаємодія клопідогрелю й аторвастатину, єдність метаболізму в печінці системою цитохром Р450, що обумовлювала взаємоослаблюючий як гіполіпідемічний, так і антитромбоцитарний ефект останніх. Ці питання і дотепер залишаються суперечливими [14].
Метою нашого дослідження стало вивчення антитромбоцитарного ефекту клопідогрелю в пацієнтів, які до розвитку ГКС з елевацією сегмента ST приймали аспірин і виявилися клінічно аспіринорезистетними. Нами обстежено 30 хворих віком 56,2 ± 3,4 року, які надходили до клініки Дорожньої клінічної лікарні № 2 м. Києва з ГІМ з елевацією сегмента ST. 15 із них до надходження до стаціонару регулярно приймали 100 мг аспірину, в анамнезі у всіх була артеріальна гіпертензія, у 70 % пацієнтів ГІМ передувала стенокардія, у 30 % хворих в анамнезі був цукровий діабет 2-го типу, ці пацієнти склали 1-шу групу. Інші на момент надходження ніякої антитромбоцитарної терапії не отримували, ці пацієнти склали 2-гу групу. Групи були порівнянними за віком, статтю, локалізацією некрозу, терапією: поряд із традиційною терапією тромболітичними засобами, гепаринами, нітратами, ІАПФ, β-адреноблокаторами, гіполіпідемічними препаратами в умовах клінічної аспіринорезистентності як у 1-й групі хворих, так і в 2-й групі призначалася з першого дня захворювання антитромбоцитарна терапія клопідогрелем (Тромбонет компанії «Фармак») у дозі 300 мг, а надалі в дозі 75 мг/добу. У 2-й групі в анамнезі була відсутня артеріальна гіпертензія, хворі мали прямі протипоказання до застосування аспірину. Контрольну групу склали 20 здорових осіб.
Вивчення гемостатичних параметрів проводилось у венозній крові двічі: до початку лікування та на 14-ту добу госпітального періоду. При цьому забір крові здійснювався з ліктьової вени і відповідав усім вимогам щодо коагулологічних досліджень.
У безтромбоцитній плазмі оцінювалися показники плазмового гемостазу, що отримували шляхом центрифугування при швидкості 3000 об./хв (1500 г) протягом 20 хв. Інформацію про стан трьох фаз згортання крові отримували за допомогою базисних коагуляційних тестів ТЧ, ПТЧ і АЧТЧ, що проводилися за стандартними коагулологічними методиками (на приладі Amelung). Вивчення фібринолітичної системи крові проводилось у двох напрямках: 1) визначення субстрату фібринолізу та продуктів його розпаду (фібриноген); 2) визначення загального фібринолітичного потенціалу (час еуглобулінового лізису, час контактного фібринолізу). Фібриноген визначався за допомогою хронометричного методу (за Клауссом), заснованого на визначенні часу згортання розведеної цитратної плазми надлишком високоактивного тромбіну. Для оцінки активності тромбоцитарного гемостазу використовували багату на тромбоцити плазму (Тп) та безтромбоцитну плазму (Бп). Тп отримували шляхом відстоювання протягом 2 год при кімнатній температурі плазми, яку відбирали з цільної крові, тим самим запобігаючи механічним травмуючим впливам на тромбоцити. Вивчення агрегаційної активності тромбоцитів здійснювалося за допомогою агрегометру виробництва фірми Solar, при цьому вивчали наявність спонтанної агрегації тромбоцитів (СА) та стимульовану агрегацію. Як індуктори агрегації застосовувались АДФ у кінцевій концентрації 1,2 × 10-6 М і ристоміцин. Усі індуктори використовувалися в низьких концентраціях. Для виведення нормативних показників гемостазу нами вивчалися зразки плазми, отриманої від 2 практично здорових осіб. Забір крові проводився після 12-годинного голодування. Тропонін Т у крові визначався ферментзв'язуючим імуносорбентним методом кількісно за допомогою приладу Cardiac Reader (Roche Diagnostics Corporation, Indianapolis, USA). Результати обстежень оброблялися шляхом визначення для кожного варіаційного ряду середньої арифметичної величини М та стандартного відхилення σ. Дані подано у форматі М ± σ. Визначення вірогідності отриманих даних встановлювалося за допомогою t-критерію Стьюдента. Розрахунки проводились на ПК за допомогою пакету програм Excel 2000. Вірогідними вважалися відмінності при значенні р < 0,05.
У 1-й групі хворих при моніторингу тромбоцитарного гемостазу ступінь агрегації тромбоцитів, ініційованої АДФ та ристоміциновими агоністами, на 14-й день вірогідно був нижчим від вихідного рівня відповідно в 2,0 (р < 0,05) і 2,1 раза, проте в останньому випадку не мав ознак вірогідності. Щодо швидкості АДФ-агрегації тромбоцитів під час проведеного лікування, то вона також у цей період обстеження вірогідно знижувалася в 1,6 раза (р < 0,05) порівняно з вихідним рівнем. Швидкість ристоміцинової агрегації вірогідно знижувалася вже з 14-го дня лікування і була в 1,8 раза нижчою порівняно з вихідною (р < 0,05), а час агрегації в 1,5 раза подовжувався (р < 0,05). На фоні лікування клопідогрелем дезагрегація при застосуванні АДФ-індуктора в 1,2 раза (р < 0,05) зменшувалася порівняно з вихідними даними (табл. 1).
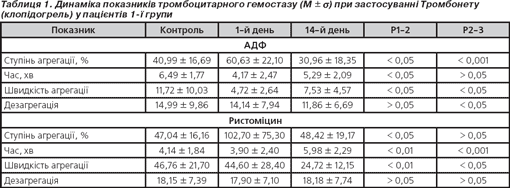
Згідно з отриманими даними встановлено, що клопідогрель із 14-го дня застосування пригнічував як ступінь, так і швидкість АДФ-агрегації тромбоцитів подібно до ристоміцинової агрегації, проте зниження швидкості агрегації тромбоцитів при застосуванні ристоміцину відзначалося більшою мірою. Це стосувалось і ступеня ристоміцинової агрегації, що більшою мірою пригнічувалася порівняно з АДФ-агрегацією, проте це не носило ознак вірогідності. Починаючи з 14-го дня вірогідне зниження швидкості ристоміцинової агрегації вказувало на непрямий зв'язок включення одного із компонентів судинного гемостазу — фактору Віллебранда (табл. 2, 3).
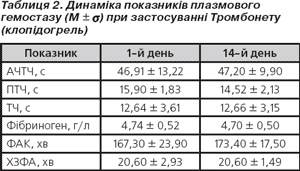
Щодо плазмового гемостазу, результати якого подані в табл. 2, то вірогідних відмінностей серед показників плазмового гемостазу при динамічному моніторингу не спостерігалося як у 1-й, так і в 2-й групі. Дані мали схожий характер. Тенденція до зниження рівня фібриногену з 7-го дня застосування клопідогрелю в 2-й групі змінювалася невірогідним підвищенням на 2-й тиждень лікування, і в абсолютних цифрах рівень фібриногену сягав вихідного. Протягом динамічного періоду спостереження відзначалася депресія ХЗФА на фоні відносної активності фібринолітичної системи.
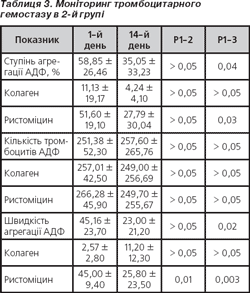
При моніторингу тромбоцитарного гемостазу 2-ї групи ступінь агрегації тромбоцитів, ініційованої АДФ та ристоміциновими агоністами, на 14-й день застосування клопідогрелю вірогідно був нижчим порівняно з вихідним рівнем в 1,6 і 1,8 раза відповідно (р < 0,05). Щодо швидкості агрегації тромбоцитів у ході проведеного лікування, то вона також у цей період обстеження вірогідно знижувалася в 1,9 раза (р < 0,02) порівняно з вихідним рівнем, і більшою мірою це стосувалось АДФ-агрегації, тоді як швидкість ристоміцинової агрегації вірогідно знижувалася вже з 7-го дня лікування і в 1,5 раза була нижчою порівняно з вихідною (р = 0,01) (табл. 3).
Що стосується плазмового гемостазу, результати якого подані в табл. 2, то вірогідних відмінностей серед показників плазмового гемостазу при динамічному моніторингу не спостерігалося. Тенденція до зниження фібриногену з 7-го дня застосування клопідогрелю змінювалася невірогідним його підвищенням на 2-й тиждень лікування, і в абсолютних цифрах рівень фібриногену сягав вихідного. Протягом динамічного періоду спостереження відзначалося пригнічення ХЗФА на фоні відносно високої фібринолітичної активності.
Висновки
Тромбонет (клопідогрель) як за умов клінічної аспіринорезистентності, так і в групі пацієнтів, хворих на ГКС з елевацією сегмента ST, які не отримували аспірин до надходження до стаціонару, вірогідно впливає на показники тромбоцитарно-судинного гемостазу. Він виявився вірогідно ефективним як у 1-й групі пацієнтів, які до розвитку ГІМ приймали аспірин, так і в 2-й групі. Уже з 2-го тижня застосування клопідогрелю відзначалося зниження більшою мірою ступеня і швидкості АДФ-агрегації тромбоцитів. Починаючи з 14-го дня застосування клопідогрелю відзначалося більш раннє сповільнення ристоміцинової швидкості й часу агрегації тромбоцитів, що вказує на опосередкований вплив клопідогрелю на судинний гемостаз, зокрема на фактор Віллебранда.
Відсутність вірогідного впливу на плазмовий гемостаз, зокрема на рівень фібриногену, вигідно вирізняє клопідогрель серед інших тієнопіридинів і дає можливість рекомендувати його в комбінації з тромболітичними препаратами, зокрема стрептокіназою, з перших днів лікування.
Належне ведення пацієнтів із ГКС з елевацією сегмента ST, асоційованим із артеріальною гіпертензією, дозволяє підвищити ефективність антитромбоцитарного лікування у пацієнтів, попередньо лікованих аспірином, шляхом збільшення дози клопідогрелю (300 мг) для запобігання феномену недостатності клопідогрелю, у тому числі не лише при артеріальній гіпертензії, а й при станах, що клінічно збільшують ризик аспіринорезистентності (надмірна вага, цукровий діабет, гіперхолестеринемія, високий рівень фібриногену, МВ КФК), а також у пацієнтів, яким здійснювався системний тромболізис, зокрема, нефібринспецифічними тромболітичними засобами. Результатом вжитих заходів є зменшення проявів синдрому рикошету і мінімізація ризику системних кровотеч.
Застосування сучасних аналогів клопідогрелю, а саме препарату Тромбонет національного виробника — компанії «Фармак», з огляду на результати проведеного дослідження є доцільним та дає можливість суттєво вплинути на частоту серйозних судинних подій та покращити результати лікування й віддалений прогноз у пацієнтів з ГІМ та елевацією сегмента ST.
1. FitzGerald G.A., Gates J.A., Hawiger J., Maas R.L., Roberts U., Lawson J.A., Brash A.R. Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man // J. Clin. Invest. — 1983. — 71. — 676-88.
2. Antithrombotic Trialists ' Collaboration. Collaborative meta-analysis of randomized trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients // BMJ. — 2002. — 324. — 71-86.
3. Catella-Lawson F., Reilly M.P., Kapoor S.C., Cucchiara A.J., DeMarco S., Tourninr B., Vyas S.N., FitzGerald G.A. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin // N. Engl. J. Med. — 2001. — 345. — 1809-17.
4. Altman R., Scazziota A. Why aspirin cannot prevent arterial thrombosis // Circulation. — 1996. — 94. — 3002-3.
5. Patrono C. Aspirin resistance: definition, mechanism and clinical read-outs // J. Thromb. Haemost. — 2003. — 1. — 1710-13.
6. Muller I., Besta F., Schuiz C., Massberg S., Schonig A., Gawaz M. Prevalence of clopidogrel non-responders among patients with stable angina pectoris scheduled for elective coronary stent placement // Thromb. Haemost. — 2003. — 89. — 783-7.
7. Halushka M.K., Halushka P.V. Why are some individuals resistant to the cardioprotective effects of aspirin? Could it be thromboxane A2? // Circulation. — 2002. — 105. — 1620-2.
8. Neumann F.J., Kastrati A., Pogatsa-Murray G., Mehilli J., Bollwein H., Bestehorn H.P., Schmitt C., Seyfarth M., Dirschinger J., Schomig A. Evaluation of prolonged antithrombotic pretreatment («cooling-off» strategy) before intervention in patients with unstable coronary syndromes: a randomized controlled trial // JAMA. — 2003. — 290. — 1593-9.
9. McKee S.A., Sane D.C., Deliargiris E.N. Аspirin resistance in cardiovascular disease: a review of prevalence, mechanism, and clinical significance // Thromb. Haemost. — 2002. — 88. — 711.
10. Fagan S.C., Simonson W. Antiplatelet therapy in the management of atherothrombosis in the elderly // Clinical Consult. — 2003. — 18. — 1-10.
11. Eikelboom J.W., Hirsh J., Weitz J.I. et al. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events // Circulation. — 2002. — 105. — 1650-1655.
12. Gurbel P.A., Bliden K.P. Durability of platelet inhibition by clopidogrel // Am. J. Cardiol. — 2003. — 91. — 1123-1125.
13. Soffer D., Moussa I., Harjai K.J. et al. Impact of angina class on inhibition of platelet aggregation following clopidogrel loading in patients undergoing coronary intervention: do we need more aggressive dosing regimens in unstable angina? // Catheter Cardiovasc. Interv. — 2003. — 59. — 21-25.
14. Lau W.C., Waskell L.A., Watkins P.B. et al. Atorvastatin reduces the ability of clopidogrel to inhibit platelet aggregation: a new drug-drug interaction // Circulation. — 2003. — 107. — 32-37.
15. Matetzky S., Shenkman B., Guetta V. et al. Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction // Circulation. — 2004. — 109. — 3171-3175.
16. Mahaffey K.W., Cohen M., Garg J., Antman E. High r isk p atients with a cute Coronary Syndrome treated with l ow m olecular Weight or Unfractioned Heparin // JAMA. — 2005. — 294. — 2594-2600.
17. Lev et al. Aspirin and Clopidogrel Drug Response in patients Undergoing Percutaneous Coronary Intervention // JACC. — 2006 — 47 (1). — 27-33.
18. FitzGerald GA. Parsing an enigma: the pharmacodynamics of aspirin resistance // Lancet. — 2003. — 361. — 542-4.
19. Awtry E.H., Loscalzo J. Aspirin // Circulation. — 2000. — 101. — 1206-18.
20. De Gaetano G., Donati M.B. Prevention on thrombosis and vascular inflammation: benefits and limitations of selective or combined COX-1, COX-2 and 5-LOX inhibitors // Trend Pharmacol. Sci. — 2003. — 24. — 245-52.
21. Syrbe G., Redlich H., Weidlich B. et al. Individual dosing of ASA prophylaxis by controlling platelet aggregation // Clin. Appl. Thromb. Hemost. — 2001. — 7. — 209-13.
22. Schafer A.I. Genetic and Acquired Determinants of Individual Variability of Response to Antiplatelet Drugs // Circulation. — 2003. — 108. — 910-1.
23. Zimmermann N., Wenk A., Kim U., Kienzle P., Weber A.A., Gams E., Schror K. Functional and Biochemical Evaluation of Platelet Aspirin Resistance аfter Coronary аrtery Bypass Surgery // Circulation. — 2003. — 108. — 542-7.
24. Sane D.C., McKee S.A., Malinin A.I., Serebruany V.L. Frequency of aspirin resistance in patients with congestive heart failure treated with antecedent aspirin // Am. J. Cardiol. — 2002. — 90. — 893-5.
25. Li N., Hu H., Hjemdahl P. Aspirin treatment does not alternate platelet or leukocyte activation as monitored by whole blood flow cytometry // J. Thromfi. Res. — 2003. — 111. — 165-170.
26. Mehta J., Mehta P., Burger C., Pepine C.J. Platelet aggregation studies in coronary artery disease. Past 4. Effect of aspirin // Atherоsclerosis. — 1978. — 31. — 169-75.
27. Koksch M., Zeiger F., Wittig K., Siegemund A., Reininger C.B., Pfeiffer D., Ruehlmann C. Coagulation, fibrinolysis and platelet P-selectin expression in peripheral vascular disease // Eur. J. Vase Endovasc. Surg. — 2001. — 21. — 147-54.
28. Andersen K., Hurlen M. Aspirin non-responsiveness as measured by PFA-100 in patients with coronary artery disease // Thromb. Res. — 2002. — 108. — 37-42.