Статья опубликована на с. 28-32 (Укр.)
Хроническая болезнь почек (ХБП) — это нарушение гомеостаза, вызванное необратимым снижением массы действующих нефронов почек, которое возникает при всех прогрессирующих заболеваниях почек и проявляется многосимптомным комплексом, отражающим участие в этом процессе практически всех органов и систем больного. ХБП определяется как повреждение почек или снижение их функции в течение 3 месяцев или более независимо от диагноза. Такое временное ограничение (критерий «стойкости») в качестве временного параметра определения ХБП было выбрано потому, что в данные сроки острые варианты развития дисфункции почек, как правило, завершаются выздоровлением. Хроническая болезнь почек является важной медицинской и социальной проблемой современности, распространенность которой достигает 5–11 % в общей популяции [1, 2]. Существенное влияние на развитие и прогрессирование хронических дисфункций почек в той или иной популяции оказывает целый ряд факторов, к которым относятся: увеличение возраста популяции, уровень заболеваемости некоторыми инфекциями, алкоголь и курение, состояние окружающей среды, климат, традиции питания, генетические особенности населения и др. Около 40 % взрослых имеют повышенный риск развития ХБП, среди которых значительное число больных с артериальной гипертензией, метаболическим синдромом и сахарным диабетом, что приводит к резкому снижению качества жизни, высокой смертности, а также к необходимости применения дорогостоящих методов заместительной терапии в терминальной стадии — диализа и пересадки почки. Увеличивающееся быстрыми темпами число больных с терминальной почечной недостаточностью (ТПН) требует постоянного увеличения расходов на проведение диализа и трансплантации почек. Несмотря на то, что только небольшая часть больных с ХБП нуждается в заместительной почечной терапии (ЗПТ), расходы на проведение ЗПТ весьма существенные и становятся обременительными даже для стран с высокоразвитой экономикой. ХБП является обобщающим термином и самостоятельным диагнозом. Помимо многообразия этиологических факторов, характерных для ХБП, большинство хронических заболеваний почек имеют единый механизм прогрессирования, а морфологические изменения в почках при почечной недостаточности однотипны. В конечном итоге они сводятся к преобладанию фибропластических процессов с замещением функционирующих нефронов соединительной тканью и сморщиванию почек, что приводит к гибели нефронов. Поэтому в настоящее время ХБП является глобальной общественной задачей.
Современные международные рекомендации предлагают классифицировать ХБП на пять стадий с учетом величины скорости клубочковой фильтрации (СКФ) [3] (табл. 1), так как СКФ имеет самостоятельное диагностическое и прогностическое значение. Кроме того, новые рекомендации предполагают разделение 3-й стадии ХБП на стадии 3а и 3б ввиду того, что почечный прогноз не одинаков в группах лиц с ХБП 3-й стадии со СКФ от 59 до 45 мл/мин/1,73 м2 и от 44 до 30 мл/мин/1,73 м2.
/28_u2.jpg)
Показатель СКФ на уровне 90 мл/мин принят как нижняя граница нормы. Значение СКФ < 60 мл/мин выбрано ввиду соответствия гибели более 50 % нефронов (рис. 1).
/28_u.jpg)
Нарушение липидного обмена при ХБП
У больных с диагнозом ХБП одним из факторов риска данного заболевания является развитие и прогрессирование нарушений липидного обмена [4–6]. Согласно многочисленным клиническим исследованиям, гиперлипидемия стоит на первом месте среди метаболических расстройств при ХБП [7].
Предположение о взаимосвязи между накоплением липидов и заболеванием почек впервые было сделано еще в 1860 г. Рудольфом Вирховым [8], который в своих лекциях в Институте патологии г. Берлина отмечал «жировое перерождение почечного эпителия как стадию болезни Брайта» (историческое обозначение гломерулонефрита, описанного в XIX веке британским ученым Ричардом Брайтом, одним из отцов-основателей нефрологии) [9].
В 1982 году в журнале Lancet впервые была опубликована статья J. Moorhead и соавт. [10], в которой авторы предложили гипотезу нефротоксичности липидов, что послужило стимулом для дальнейшего исследования липидов при болезни почек. Это была первая публикация, где введено понятие о том, что компенсационный синтез липопротеидов печени в ответ на экскрецию альбумина с мочой может привести к прогрессивным заболеваниям почек и что патогенез атеросклероза и гломерулосклероза при повреждении почек может иметь общий путь. При этом процессе персистирующая альбуминурия стимулирует избыток синтеза липопротеидов в печени, тем самым нарушая цикл синтеза липидов. Было высказано предположение, что многие из заболеваний гломерулярного и тубулоинтерстициального аппарата связаны с атеросклерозом (предложен термин «гломерулярный атеросклероз»), в том числе с дислипидемиями. С тех пор многочисленные клинические и лабораторные исследования подтвердили гипотезу о том, что гиперлипидемия является результатом компенсаторного синтеза липопротеидов печени в ответ на экскрецию альбумина с мочой и способствует прогрессированию атеросклероза и гломерулосклероза [11].
Основные классы липидов и липопротеидов (ЛП) плазмы крови
Основными липидами, находящимися в плазме крови человека, являются:
I. Липиды:
— холестерин (ХС);
— триглицериды (триацилглицериды) (ТГ);
— фосфолипиды (ФЛ);
— жирные кислоты (ЖК).
II. Липопротеины:
— хиломикроны (ХМ);
— липопротеины очень низкой плотности (ЛОНП);
— липопротеины промежуточной плотности (ЛППП);
— липопротеины низкой плотности (ЛПНП);
— липопротеины высокой плотности (ЛПВП).
В клинической практике принято оценивать содержание липидов только в плазме или сыворотке крови. Их расчет не производится на объем цельной крови. В сыворотке определяется в среднем на 3 % больше ХС и ТГ, чем в плазме. Для особо точных измерений и сопоставлений используется следующая формула: ХСплазмы = ХСсыворотки : 1,03. Для ТГ применяется аналогичная формула.
Уровень липидов в крови зависит от возраста, половой принадлежности, факторов внешней и внутренней среды, например характера питания, физической активности, гормонального статуса и др.
I. Липиды
1. Холестерин
Холестерин представляет собой вторичный одноатомный ароматический спирт (рис. 2). Он обнаруживается во всех тканях и жидкостях человеческого организма как в свободном состоянии, так и в виде сложных эфиров. Является важным компонентом клеточных мембран, служит предшественником образования стероидных гормонов и желчных кислот. Около 70 % холестерина в липопротеидах плазмы крови представлено эфирами холестерина. У практически здоровых людей 2/3 холестерина плазмы содержится в составе атерогенных, 1/3 — антиатерогенных липопротеидов. Около 80 % эндогенного ХС вырабатывается самим организмом (печенью, кишечником, почками, надпочечниками, половыми органами), а 20 % экзогенного ХС поступает в организм в составе пищи.
2. Триглицериды (триацилглицериды)
Триглицериды — сложные эфиры глицерина и высших жирных кислот (стеариновой, пальмитиновой и др.) (рис. 3). Нейтральный жир, поступающий с пищей, гидролизуется в просвете тонкого кишечника, а продукты распада (глицерин и жирные кислоты) используются в клетках слизистой оболочки тонкого кишечника для ресинтеза триацилглицеридов, которые включаются в состав хиломикронов.
3. Фосфолипиды
Фосфолипиды представляют собой соединение спирта глицерола с высшими жирными кислотами и фосфорной кислотой, а также азотсодержащие соединения — холин, этаноламин, серин, инозитол, которые входят в состав ХМ и ЛП (рис. 4). ФЛ играют важную роль в структуре и функции клеточных мембран, в активации мембранных и лизосомальных ферментов, проведении нервных импульсов, свертывании крови, иммунологических реакциях, процессах клеточной пролиферации и регенерации тканей, переносе электронов в реакциях дыхательной цепи. Особая роль принадлежит ФЛ в формировании липопротеиновых комплексов.
Состоит из глицеринового фрагмента, двух жирных кислот, фосфатной группы и полярной молекулы.
4. Жирные кислоты
Синтез ЖК происходит в печени, стенке кишечника, легочной ткани, жировой ткани, в ткани мозга, почках, костном мозге, сосудистой стенке и протекает в цитозоле клетки. Характерно, что в цитозоле печеночных клеток синтезируется главным образом пальмитиновая кислота. Другие ЖК синтезируются в печени путем удлинения (элонгации) цепи на основе уже синтезированной пальмитиновой кислоты (рис. 5).
II. Липопротеины
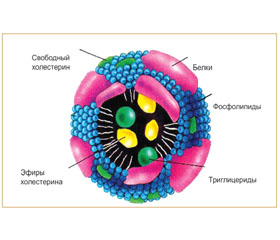
Все липиды плазмы крови находятся в составе липопротеидов (рис. 6). Необходимо отметить, что при анализе липидов в плазме (сыворотке) крови фактически определяется их суммарное содержание во всех классах ЛП.
Плазменные липопротеиновые частицы имеют сферическую форму. Внутри находится жировая «капля», содержащая неполярные липиды (ТГ и эфиры ХС), которые формируют ядро липопротеиновой частицы. Основную массу липопротеиновой частицы составляет ее ядро, окруженное оболочкой из фосфолипидов, холестерина и белка. В зависимости от класса ЛП изменяется соотношение между основными липидами: с увеличением плотности частиц уменьшается доля ТГ и возрастает доля эфиров ХС (ЭХС) (рис. 7).
Липопротеины имеют важное клиническое значение для организма человека (табл. 2).III. Аполипопротеины
Аполипопротеины (апопротеины, апо) являются белковыми компонентами ЛП. Они выполняют не только структурную функцию, но и обеспечивают активное участие ЛП-комплексов в транспорте липидов в кровотоке от мест их синтеза к клеткам периферических тканей, а также в обратном транспорте ХС в печень для дальнейшего метаболизма. Аполипопротеины выполняют функцию лигандов во взаимодействии ЛП со специфическими рецепторами на клеточных мембранах, регулируя тем самым гомеостаз ХС в клетках и в организме в целом. Участвуют в регуляции активности ряда ферментов липидного обмена: лецитин-холестерин-ацилтрасферазы (ЛХАТ), липопротеинлипазы (ЛПЛ), печеночной триацилглицеринлипазы (П-ТГЛ).
Апопротеины семейства А (апоА-I и апоА-II)
находятся в крови человека в довольно высоких концентрациях. По абсолютному содержанию апоА-I занимает первое, а апоА-II — третье место среди всех апопротеинов (второе место принадлежит белку В-100).
Апопротеин А-I представляет собой полипептид, содержащий 245 аминокислотных остатков с молекулярной массой 28 кДа. Его содержание составляет около 70 % общей массы белка в ЛПВП, что указывает на его важную структурную роль.
Апопротеин А-II занимает второе место после апоА-I среди апопротеинов ЛПВП, на его долю приходится около 20 % всех белков частицы. АпоА-II состоит из двух одинаковых полипептидных цепей. Каждая цепь образована 77 аминокислотными остатками и имеет молекулярный вес 86 кДа. Синтез апоА-II происходит в стенке тонкой кишки и печени. Белок поступает в кровь в составе ХМ и ЛПВП. АпоА-II выполняет структурную функцию. Метаболическая функция этого белка состоит в ингибировании ЛХАТ и активировании ТГЛ.
У здорового человека содержание липидов находится в пределах определенных физиологических колебаний, которые принимаются за норму. В практической медицине для липидного профиля используют определение общего холестерина, холестерина ЛПНП, холестерина ЛПВП, триглицеридов (табл. 3).
Для определения оптимального значения уровня липидов в плазме используется «Правило 5» (табл. 4).
Хроническая болезнь почек, связанная с патологией обмена липидов
Гиперлипидемия может реализовать свое влияние на прогрессирование почечного повреждения несколькими путями.
1. Путем развития интраренального атеросклероза.
2. Через токсическое влияние липидов на структуры нефрона.
Основные механизмы прогрессирования ХБП, связанные с обменом липидов, отличаются в зависимости от стадии процесса. При этом имеются некоторые общие черты развития, в основе которых лежат повышенные уровни ХС, ТГ, ЛПНП и низкий уровень ЛПВП в плазме крови [12, 13]. Было показано, что у нефрологических пациентов дислипидемия приводит к повреждению эндотелия капилляров клубочков и отложению липидов в мезангиальных клетках, которые связывают и окисляют ЛПНП, стимулируя пролиферацию мезангия и развитие гломерулосклероза [14]. Гиперлипидемия повышает активацию мезангиальных клеток, имеющих рецепторы к ЛПНП, что приводит к стимуляции клеточной пролиферации и увеличению синтеза макрофагов, факторов хемотаксиса, компонентов внеклеточного матрикса, активатора плазминогена-1, активных форм кислорода и др. [15–17]. При этом ЛП, отложившиеся в базальной мембране клеток, связывают гликозаминогликаны и тем самым повышают проницаемость мембраны для белков. В результате этого процесса отфильтрованные в клубочках ЛП оседают в канальцах почек, что инициирует тубулоинтерстициальные процессы и склероз. В дальнейшем повышенное содержание липидов приводит к захвату их эпителием канальцев и депозиции внутри клеток. Отложение липидов в мезангиоцитах и канальцевом эпителии придает клеткам характерный пенистый вид. Это приводит к их дистрофии и атрофии с накоплением липидного материала в межклеточном пространстве [18] (рис. 8).
/31_u.jpg)
Морфологическим субстратом ХБП является гломерулосклероз, характеризующийся независимо от первичной патологии почек склерозом мезангия, экспансией внеклеточного матрикса, в состав которого входят ламинин, фибронектин, гепарансульфат протеогликан, коллаген IV типа и интерстициальный коллаген (в норме отсутствует в клубочках). Увеличение внеклеточного матрикса, замещающего функционально активную ткань, — комплексный процесс, протекающий с участием различных факторов роста, цитокинов и тепловых шоковых протеинов. Установлено, что у большинства больных с СКФ около 25 мл/мин и ниже терминальная хроническая почечная недостаточность возникает независимо от характера заболевания. Существует адаптивный ответ внутрипочечной гемодинамики на потерю массы действующих нефронов. Это проявляется в снижении сопротивления в афферентной и эфферентной артериолах функционирующих нефронов, приводящем к усилению скорости внутриклубочкового плазмотока, то есть к гиперперфузии клубочков и повышению гидравлического давления в их капиллярах. В результате возникает гиперфильтрация, а впоследствии — гломерулосклероз. Дисфункция эпителия канальцев тесно связана с развитием тубулоинтерстициального фиброза (рис. 9). Канальцевый эпителий способен к синтезу широкого спектра цитокинов и факторов роста. В ответ на повреждение или перегрузку он усиливает экспрессию молекул адгезии, синтез эндотелина и других цитокинов, способствующих тубулоинтерстициальному воспалению и склерозу. Любое повреждение стенки сосуда стимулирует агрегацию тромбоцитов с выбросом тромбоксана — мощного вазоконстриктора, играющего интегральную роль в развитии артериальной гипертензии. Усиление реактивности и агрегации тромбоцитов стимулирует гиперлипидемия, сочетание которой с артериальной гипертензией сопровождается еще более выраженными изменениями клубочков.
Метаболизм основных липидов при ХБП
1. Общий холестерин при ХБП
У пациентов с ХБП характер дислипидемии отличается в зависимости от стадии процесса [21]. Считается, что наибольшее повреждение клубочков почек вызывает высокий уровень общего холестерина сыворотки. Гиперхолестериновая диета у экспериментальных животных вызывает появление в клубочках липидных депозитов, моноцитарной инфильтрации и гиперклеточности мезангия, а также увеличение мезангиального матрикса. Одновременно с увеличением уровня общего холестерина нарастает протеинурия и количество склерозированных клубочков. Даже просто гиперхолестеринемия приводит к развитию протеинурии, уремии и гломерулосклерозу и увеличению внутриклубочкового давления. Клиническими исследованиями показано, что гиперлипидемия при любых нефропатиях ускоряет прогрессирование почечной недостаточности. При этом скорость прогрессирования зависит от уровня общего холестерина сыворотки.
2. Триглицериды при ХБП
Среди возможных механизмов уремической гипертриглицеридемии обсуждают различные процессы: изменение активности ферментов, нарушения в рецепторном аппарате, снижение метаболизма ЛПВП. Было показано, что уже на ранних стадиях ХБП повышается уровень триглицеридов плазмы крови, достигая максимальных значений у больных с нефротическим синдромом и у пациентов, получающих заместительную почечную терапию [22]. Это происходит за счет уменьшения активности таких ферментов, как липопротеинлипаза (ЛПЛ) (КФ 3.1.1.34) и печеночная триацилглицеринлипаза (П-ТГЛ) (КФ 3.1.1.3).
Механизм действия липопротеинлипазы заключается в расщеплении ТГ самых крупных по размеру и богатых липидами липопротеинов плазмы крови — ХМ и ЛПОНП (рис. 10). Хиломикроны, циркулирующие в крови, постепенно высвобождаются от триацилглицеринов (в результате контактов с липопротеинлипазой) и превращаются в остаточные хиломикроны, которые содержат очень мало триацилглицеринов и много холестерина. Остаточные хиломикроны (частично и цельные) поглощаются клетками печени.
Печеночная триглицеринлипаза катализирует гидролиз ТГ в ЛППП, в результате которого образуются ЛПНП. Гидролитическое действие фермент осуществляет на эндотелиальной поверхности капилляров печени во время прохождения через них крови (это определило название фермента). Гидролиз триацилглицеринов катализируется тремя липазами: триацилглицерин-, диацилглицерин- и моноацилглицеринлипазой. Активность двух последних ферментов в 10–100 раз превышает активность триацилглицеринлипазы, которая является регуляторным ферментом. Действие триацилглицеринлипазы состоит в расщеплении триацилглицеринов с образованием 1,2-диацилглицерина и свободных жирных кислот (рис. 11).
/31_u2.jpg)
В результате уменьшения активности данных ферментов происходит накопление ТГ в составе ХМ и ЛПОНП. Из-за снижения активности липаз нарушается расщепление ТГ до свободных жирных кислот, необходимых для обеспечения энергетических потребностей организма, что может оказывать влияние на развитие синдрома белково-энергетической недостаточности у больных, находящихся на гемодиализе [23].
3. Липопротеины высокой плотности при ХБП
Характерным для ХБП является снижение концентрации ЛПВП. Этому способствует низкая концентрация и активность лецитин-холестерин-ацилтрансферазы, что приводит к нарушению синтеза, транспорта ЛПВП и быстрому расщеплению ЛПВП [22, 24]. На более поздних стадиях, но еще в додиализном периоде, у пациентов обычно имеются повышенные уровни ЛПНП и низкий уровень ЛПВП. У пациентов со значительной протеинурией и нефротическим синдромом нарушения липидного обмена также выражены за счет повышения ЛПНП, гипертриглицеридемии и гиперхолестеринемии. Гипоальбуминемия, часто сопутствующая терминальной почечной недостаточности и ХБП, также потенциально может способствовать снижению содержания ЛПВП. Объясняется это тем, что ЛПВП получают существенное количество холестерина от альбумина, который выступает в качестве переносчика свободного холестерина из периферических тканей к ЛПВП, в то же время происходит образование дефектных окисленных форм ЛПВП, которые, в свою очередь, приобретают прооксидантные и провоспалительные свойства. Кроме того, со степенью гипоальбуминемии обратно коррелирует выраженность гиперхолестеринемии, что объясняется компенсаторным характером повышения синтеза ЛП в печени при нефротическом синдроме. Имеет значение также пониженный катаболизм ЛП из-за уменьшения активности липопротеинлипаз.
4. Липопротеины низкой плотности при ХБП
Мезангиальные клетки, имеющие рецепторы к липопротеидам низкой плотности, связывают и окисляют их, это запускает каскад выработки цитокинов, стимулирующих пролиферацию мезангия и развитие гломерулосклероза. Параллельно снижается выработка защитных протеогликанов и коллагенолитических ферментов, регулирующих образование мезангиального матрикса, ослабляются фагоцитарные свойства мезангиоцитов, мезангий «перегружается» макромолекулами. Липопротеины, отложившиеся в базальной мембране клеток, связывают отрицательно заряженные гликозаминогликаны и нейтрализуют ее отрицательный заряд, повышая проницаемость мембраны для белков. Помимо этого, фильтрующиеся в клубочках ЛП, осаждаясь в канальцах почек, индуцируют и тубулоинтерстициальные процессы, склероз интерстиция и развитие почечной недостаточности. Современные рекомендации предлагают в качестве целевых уровни ХС ЛПНП < 2,5 ммоль/л для больных с ХБП со СКФ 30–60 мл/мин/1,73 м2 и < 1,8 ммоль/л — для больных ХБП со СКФ < 30 мл/мин/1,73 м2.
5. Аполипопротеины А-I и А-II при ХБП
Аполипопротеины А-I и А-II входят в состав ЛПВП. АпоА-I является активатором ЛХАТ и лигандом к рецепторам ЛПВП, а апоА-II — активатором печеночной липазы. У больных ТПН концентрации этих апопротеинов значительно снижены. ЛХАТ служит основной детерминантой образования и содержания ЛПВП в плазме крови. У пациентов с ТПН ее активность снижена. Дефицит данного фермента может приводить к снижению уровня ЛПВП в плазме и нарушениям превращения ЛПВП при ХБП, что сопровождается значительным повышением свободного сывороточного ХС и снижением концентрации эстерифицированного ХС (рис. 12).
6. Перекисное окисление липидов при ХБП
Активация перекисного окисления липидов в мембранах эндотелиальных структур клеток приводит к потере их функциональной активности и является одним из механизмов развития заболеваний почек, что определяет степень интоксикации при ХБП [19]. На стадии выраженной ХБП липопротеины подвергаются модификации и становятся окисленными ЛПНП (ок-ЛПНП). Ок-ЛПНП способствуют адгезии моноцитов к эндотелию капилляров клубочка и оказывают влияние на клетки канальцевого эпителия [20]. Они связываются с рецепторами в мезангии и через ряд клеточно-молекулярных механизмов усиливают в нем воспалительные и фиброгенные процессы. Цитотоксический эффект ок-ЛПНП проявляется в индукции апоптоза подоцитов с потерей нефрина и повреждением гломерулярного барьера.
Особенности коррекции липидных нарушений при ХБП
Гиполипидемическая терапия у больных ХБП на сегодняшний день является важнейшим элементом нефропротективной стратегии, призванной не только предупреждать, но и тормозить прогрессирование нефросклероза, предотвращая или откладывая развитие почечной недостаточности. Принципы лекарственной терапии являются общими для гиперлипидемий любой этиологии, причем принципиальным является начало лечения уже на ранних стадиях ХБП.
К липидкорригирующим медикаментозным средствам относятся статины, фибраты, никотиновая кислота, секвестранты желчных кислот, омега-3 полиненасыщенные жирные кислоты, антиоксиданты.
Статины являются структурными ингибиторами фермента гидрокси-метилглутарил-КоА-редуктазы (ГМГ-КоА-редуктазы) — основного фермента, регулирующего биосинтез холестерина в гепатоцитах. В результате снижения внутриклеточного содержания холестерина печеночная клетка увеличивает количество мембранных рецепторов к ЛПНП на своей поверхности. Рецепторы «распознают», связывают и выводят из кровотока атерогенные частицы ЛПНП и таким образом снижают концентрацию ХС в крови. Наряду с гиполипидемическим действием статины обладают плейотропными эффектами. В частности, они улучшают функцию эндотелия, снижают уровень С-реактивного протеина — маркера воспалительной реакции в сосудистой стенке, подавляют агрегацию тромбоцитов, ослабляют пролиферативную активность гладкомышечных клеток сосудистой стенки. Статины различают по способу их получения. Так, ловастатин, симвастатин и правастатин являются природно-синтезированными соединениями, получаемыми из продуктов жизнедеятельности некоторых видов грибков, в то время как флувастатин, аторвастатин и розувастатин являются синтезированными препаратами.
В Украине зарегистрированы оригинальные статины: ловастатин (мевакор), правастатин (липостат), флувастатин (лескол форте), симвастатин (зокор), аторвастатин (липримар) и розувастатин (крестор).
Статины наиболее эффективно снижают уровень ХС ЛПНП. Действие статинов на уровень ХС ЛПНП является дозозависимым. Каждое удвоение дозы статина приводит к дополнительному снижению уровня ХС ЛПНП на 6 % («правило 6 %»). Статины в незначительной степени влияют на уровни ТГ и ХС ЛПВП. Как правило, они снижают уровень ТГ на 10–15 % и повышают уровень ХС ЛПВП на 8–10 %.
Эти препараты не только способствуют нормализации липидного профиля и тем самым препятствуют развитию атеросклероза, но и, уменьшая накопление липидов в ткани почек, угнетают пролиферацию мезангиальных клеток и развитие гломерулосклероза.
Кроме статинов, актуальной является моно- и комбинированная гиполипидемическая терапия с использованием препаратов разных групп.
Согласно клиническому руководству по управлению ХБП в условиях дислипидемии, рекомендуется специальный подход к фармакологической коррекции нарушений липидного обмена [25, 26] (табл. 5).
В 2013 г. (KDIGО Clinical Practice Guideline for Lipid Management in Chronic Kidney Disease) были представлены рекомендации по лечению нарушений липидного обмена при ХБП [27] (табл. 6).
Таким образом, дислипидемия тесно связана с прогрессированием ХБП. Ее влияние обусловлено как атеросклеротическим поражением почечных сосудов, так и прямым нефротоксическим действием липидов. Гиполипидемическая терапия у больных с ХБП преследует главную цель — предупреждение развития и прогрессирования собственно ХБП.
Выявление начальных проявлений нарушения липидного обмена у пациентов с хроническими почечными заболеваниями позволяет определить группы высокого риска с неблагоприятным исходом ХБП, а своевременно назначенная терапия — предупредить развитие заболевания.
Конфликт интересов: не заявлен.
Список литературы
1. El Nahas A.M., Bello A.K. Chronic kidney disease: The global challenge // Lancet. — 2005. — № 365. — Р. 331-40.
2. Levey A.S., Atkins R., Coresh J. et al. Chronic kidney disease as a global public health problem: Approaches and initiatives — A position statement from Kidney Disease Improving Global Outcomes // Kidney Int. — 2007. — № 72. — Р. 247-59.
3. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Proctice Guideline for the Evaluation and Management of Chronic Kidney Disease // Kidney Int. — 2013. — 3(Suppl.). — Р. 1-150.
4. Gröne E.F., Gröne H.J. Does hyperli pidemia injure the kidney? Nature clinical practice // Nephrology. — 2008. — № 4(8). — Р. 424-5.
5. Sarnak M.J., Levey A. Cardiovascular disease and chronic renal disease: a new paradigm // Am. J. Kidney Dis. — 2000. — № 35(4). — S117-131.
6. Keane W.F. Lipids and the kidney // Kidney international. — 1994. — № 46(3). — Р. 910-20.
7. Tareeva I.E., Kutyrina M.I., Nikolaev A.Yu. The braking of chronic renal failure // Therapeutic Archives. — 2000. — № 6. — Р. 9-14.
8. Virchow R., Chance F. Cellular pathology, as based on physiological and pathological histology // J. Churchill. — London, 1860. A more precise account of fatty metamorphosis. — Р. 351.
9. Jay V. Richard Bright: physician extraordinaire // Arch. Pathol. Lab. Med. — 2000. — № 124. — Р. 1262-1263.
10. Moorhead J.F., Chan M.K., El-Nahas M., Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease // Lancet. — 1982. — № 2. — Р. 1309-11.
11. Grone H.J., Walli A., Grone E., Niedmann P., Thiery J., Seidel D., Helmchen U. Induction of glomerulosclerosis by dietary lipids. A functional and morphologic study in the rat // Lab. Invest. — 1989. — № 60. — Р. 433-46.
12. Vaziri N.D., Norris K. Lipid disorders and their relevance to outcomes in chronic kidney disease // Blood Purif. — 2011. — № 31(1–3). — Р. 189-96.
13. Chen S.C., Hung C.C., Kuo M.C. et al. Association of dyslipidemia with renal outcomes in chronic kidney disease // PLoS One. — 2013. — № 8(2). — Е55643.
14. Urazlina C.E., Zhdanova T.V., Nazarov A.V. et al. Lipid metabolism in patients with chronic renal failure // Ural Medical Journal. — 2011. — № 2(80). — Р. 122-6.
15. Rovin B.H., Tan L.C. LDL stimulates mesangial fibronectin production and chemoattractant expression // Kidney international. — 1993. — № 43. — Р. 218-25.
16. Keane W.F., O’Donnell M.P., Kasiske B.L. Oxidative modification of low-density lipoproteins by mesangial cells // Journal of the American Society of Nephrology. — 1993. — № 4. — Р. 187-94.
17. Kume S., Uzu T., Araki S. et al. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet // Journal of the American Society of Nephrology. — 2007. — № 18. — Р. 2715-23.
18. Colina I.B. Hyperli pidemia in patients with chronic kidney disease: features and approaches to treatment // The attending physician. — 2012. — № 1. — Р. 63-70.
19. Colina I.B., Stavrovskaya U., Shilov E.M. Dysli pidemia, and chronic progressive renal disease // Ter. archive. — 2004. — № 76(9). — Р. 75-8.
20. Abrass C.K. Cellular lipid metabolism and the role of lipids in progressive renal disease // Am. J. Nephrol. — 2004. — № 24. — Р. 46-53.
21. Vaziri N.D. Dyslipidemia of chronic renal failure: the nature, mechanisms, and potential consequences // Am. J. Physiol. Renal. Physiol. — 2006. — № 290. — F262-F272.
22. Bhowmik D., Tiwari S.C. Metabolic syndrome and chronic kidney disease // Indian J. Nephrol. — 2008. — № 18(1). — Р. 1-4.
23. Trevisan R., Dodesini A.R., Lepore G. Lipids and renal disease // J. Am. Soc. Nephrol. — 2006. — № 17. — S145-S147.
24. Kuznetsova E.B., Zhdanov T.V., Sadykova Yu.R. et al. Metabolic syndrome in nephrology patients // Ural Medical Journal. — 2011. — № 4. — Р. 34-41.
25. KDIGO Clinical Practice Guideline for Lipid Management in Chronic Kidney Disease // Kidney International. — 2013. — 3(Suppl.). — Р. 268-70.
26. National Kidney Foundation. KDOQI Clinical Practice Guidline for Diabetes and CKD: 2012 update // Am. J. Kidney Dis. — 2012. — № 60(5). — Р. 850-886.
27. Kidney Disease: Improving Global Outcomes (KDIGO) Lipid Work Group. KDIGO Clinical Practice Guidline for Lipid Management in Chronic Kidney Disease // Kidney Int. — 2013. — 3(Suppl.). — Р. 259-305.
1. El Nahas A.M., Bello A.K. Chronic kidney disease: The global challenge // Lancet. — 2005. — № 365. — Р. 331-40.
2. Levey A.S., Atkins R., Coresh J. et al. Chronic kidney disease as a global public health problem: Approaches and initiatives — A position statement from Kidney Disease Improving Global Outcomes // Kidney Int. — 2007. — № 72. — Р. 247-59.
3. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Proctice Guideline for the Evaluation and Management of Chronic Kidney Disease // Kidney Int. — 2013. — 3(Suppl.). — Р. 1-150.
4. Gröne E.F., Gröne H.J. Does hyperli pidemia injure the kidney? Nature clinical practice // Nephrology. — 2008. — № 4(8). — Р. 424-5.
5. Sarnak M.J., Levey A. Cardiovascular disease and chronic renal disease: a new paradigm // Am. J. Kidney Dis. — 2000. — № 35(4). — S117-131.
6. Keane W.F. Lipids and the kidney // Kidney international. — 1994. — № 46(3). — Р. 910-20.
7. Tareeva I.E., Kutyrina M.I., Nikolaev A.Yu. The braking of chronic renal failure // Therapeutic Archives. — 2000. — № 6. — Р. 9-14.
8. Virchow R., Chance F. Cellular pathology, as based on physiological and pathological histology // J. Churchill. — London, 1860. A more precise account of fatty metamorphosis. — Р. 351.
9. Jay V. Richard Bright: physician extraordinaire // Arch. Pathol. Lab. Med. — 2000. — № 124. — Р. 1262-1263.
10. Moorhead J.F., Chan M.K., El-Nahas M., Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease // Lancet. — 1982. — № 2. — Р. 1309-11.
11. Grone H.J., Walli A., Grone E., Niedmann P., Thiery J., Seidel D., Helmchen U. Induction of glomerulosclerosis by dietary lipids. A functional and morphologic study in the rat // Lab. Invest. — 1989. — № 60. — Р. 433-46.
12. Vaziri N.D., Norris K. Lipid disorders and their relevance to outcomes in chronic kidney disease // Blood Purif. — 2011. — № 31(1–3). — Р. 189-96.
13. Chen S.C., Hung C.C., Kuo M.C. et al. Association of dyslipidemia with renal outcomes in chronic kidney disease // PLoS One. — 2013. — № 8(2). — Е55643.
14. Urazlina C.E., Zhdanova T.V., Nazarov A.V. et al. Lipid metabolism in patients with chronic renal failure // Ural Medical Journal. — 2011. — № 2(80). — Р. 122-6.
15. Rovin B.H., Tan L.C. LDL stimulates mesangial fibronectin production and chemoattractant expression // Kidney international. — 1993. — № 43. — Р. 218-25.
16. Keane W.F., O’Donnell M.P., Kasiske B.L. Oxidative modification of low-density lipoproteins by mesangial cells // Journal of the American Society of Nephrology. — 1993. — № 4. — Р. 187-94.
17. Kume S., Uzu T., Araki S. et al. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet // Journal of the American Society of Nephrology. — 2007. — № 18. — Р. 2715-23.
18. Colina I.B. Hyperli pidemia in patients with chronic kidney disease: features and approaches to treatment // The attending physician. — 2012. — № 1. — Р. 63-70.
19. Colina I.B., Stavrovskaya U., Shilov E.M. Dysli pidemia, and chronic progressive renal disease // Ter. archive. — 2004. — № 76(9). — Р. 75-8.
20. Abrass C.K. Cellular lipid metabolism and the role of lipids in progressive renal disease // Am. J. Nephrol. — 2004. — № 24. — Р. 46-53.
21. Vaziri N.D. Dyslipidemia of chronic renal failure: the nature, mechanisms, and potential consequences // Am. J. Physiol. Renal. Physiol. — 2006. — № 290. — F262-F272.
22. Bhowmik D., Tiwari S.C. Metabolic syndrome and chronic kidney disease // Indian J. Nephrol. — 2008. — № 18(1). — Р. 1-4.
23. Trevisan R., Dodesini A.R., Lepore G. Lipids and renal disease // J. Am. Soc. Nephrol. — 2006. — № 17. — S145-S147.
24. Kuznetsova E.B., Zhdanov T.V., Sadykova Yu.R. et al. Metabolic syndrome in nephrology patients // Ural Medical Journal. — 2011. — № 4. — Р. 34-41.
25. KDIGO Clinical Practice Guideline for Lipid Management in Chronic Kidney Disease // Kidney International. — 2013. — 3(Suppl.). — Р. 268-70.
26. National Kidney Foundation. KDOQI Clinical Practice Guidline for Diabetes and CKD: 2012 update // Am. J. Kidney Dis. — 2012. — № 60(5). — Р. 850-886.
27. Kidney Disease: Improving Global Outcomes (KDIGO) Lipid Work Group. KDIGO Clinical Practice Guidline for Lipid Management in Chronic Kidney Disease // Kidney Int. — 2013. — 3(Suppl.). — Р. 259-305.

/29_u.jpg)
/29_u2.jpg)
/29_u3.jpg)
/30_u.jpg)
/32_u.jpg)