
Международный эндокринологический журнал 8 (56) 2013
Вернуться к номеру
Цукровий діабет: діагностичні критерії, етіологія і патогенез
Авторы: Панькaів В.І. - Український науково-практичний центр ендокринної хірургії, трансплантації ендокринних органів і тканин МОЗ України, м. Київ
Рубрики: Эндокринология
Разделы: Медицинское образование
Версия для печати
Статья опубликована на с. 53-64
Критерії діагностики цукрового діабету
Обстеження на цукровий діабет 2-го типу, а також діагностування захворювання проводиться шляхом визначення рівня глікемії (табл. 1). Раннє виявлення ЦД 2-го типу в пацієнтів без симптомів дозволяє своєчасно виявити захворювання, запобігти ускладненням або відстрочити їх виникнення. Правильне встановлення діагнозу ЦД 2-го типу та своєчасне лікування дозволяє усунути або зменшити симптоми захворювання та відтермінувати розвиток ускладнень.
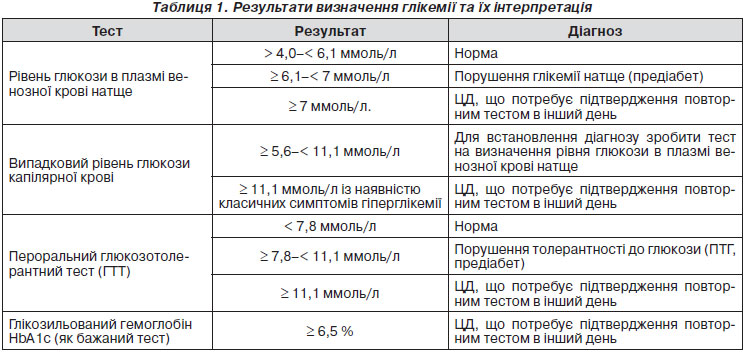
Визначення показника глікемії проводиться шляхом встановлення рівня глюкози капілярної крові в будь-який час доби незалежно від прийому їжі та/або встановлення рівня глюкози у плазмі венозної крові натще (після попереднього 8-годинного голодування).
Діагноз ЦД установлюється за наявності симптомів глікемії (спрага, часте сечовиділення, порушення зору, апатія, схуднення) та підвищення одного з показників глікемії понад зазначений рівень. За відсутності симптомів і підвищення одного з результатів глікемії понад зазначений рівень тестування проводиться в інший день.
На сьогодні не рекомендується для встановлення діагнозу ЦД використання як вимірювального приладу портативних глюкометрів та тест-смужок.
Обстеження на виявлення ЦД 2-го типу слід проводити щороку в пацієнтів із предіабетом, за наявності в пацієнта будь-якого віку надмірної маси тіла або ожиріння та одного або більше додаткових факторів ризику ЦД 2-го типу, пацієнтам із помірним, високим та дуже високим ризиком ЦД 2-го типу, усім пацієнтам після 45 років. Якщо показники рівня глюкози в межах норми, рекомендується проведення повторного тесту не пізніше як через 3 роки (або частіше, якщо виникає така необхідність).
Алгоритм проведення глюкозотолерантного тесту
Упродовж трьох діб перед проведенням тесту харчування людини має звичайний характер, з достатньою кількістю вуглеводовмісних продуктів. За 3 доби відміняється прийом тіазидних діуретиків, контрацептивних препаратів, глюкокортикоїдів. При систематичному застосуванні будь-яких препаратів необхідно повідомити лікаря. Тест проводиться вранці натще після 12-годинного попереднього голодування.
У день проведення тесту визначається рівень глікемії натще. Обстежуваний упродовж 5 хвилин приймає 75 г глюкози, розчиненої у 250–300 мл води. Під час проведення тесту виключаються підвищене фізичне навантаження, паління, вживання їжі. Рівень глікемії визначається через 1 і 2 год після прийому глюкози.
Дослідження рівня глікемії з метою виявлення ЦД не проводиться у гострій стадії хвороби або на тлі загострення хронічного захворювання, при травмах, у пацієнтів із гострим цирозом печінки, у період проведення курсового лікування препаратами, що мають гіперглікемізуючі властивості (глюкокортикостероїди, тіазидні діуретики, бета-адреноблокатори тощо).
Діагностика ЦД ґрунтується на зіставленні клінічних симптомів і лабораторних маркерів змін глікемії. До проявів гіперглікемії належать поліурія, полідипсія, втрата маси тіла (іноді з поліфагією), затуманення зору. В осіб похилого віку маніфестація ЦД може бути безсимптомною.
ЦД 2-го типу має довгу безсимптомну доклінічну стадію, що доволі часто не розпізнається, тому до моменту встановлення діагнозу понад 50 % пацієнтів мають одне або декілька ускладнень. Актуальність проблеми скринінгу ЦД 2-го типу та його ускладнень визначається ще й труднощами його виконання. Протягом тривалого часу людина може не підозрювати про наявність ЦД. У 50 % випадків ЦД 2-го типу виявляється на 5–7-му році від початку захворювання, тому у 20–30 % цих хворих у момент виявлення діабету вже діагностуються і специфічні для нього ускладнення — катаракта, ретинопатія, нефропатія, нейропатія, синдром діабетичної стопи, ішемічна хвороба серця (ІХС), артеріальна гіпертензія (АГ) тощо.
Одночасно із скринінгом ЦД 2-го типу здійснюється і скринінг інших компонентів метаболічного синдрому (МС), що також полягає в оцінці ступеня ризику захворювання. Незалежно від наявності або відсутності МС пацієнти піддаються подальшому скринінгу на ЦД — визначенню глікемії. Необхідність одночасного скринінгу на МС пояснюється тим, що за відсутності ЦД людина йде з упевненістю, що все добре, і втрачає і без того низьку мотивацію на зміну способу життя.
Сьогодні особлива увага звертається на визначення глікозильованого гемоглобіну (HbA1c). Перевага визначення HbAlc полягає в тому, що тест може бути проведений у будь-який час на відміну від ГТТ (натще, 2 години очікування, потреба придбати 75 г порошку глюкози), відзначається менша варіабельність значень у різні дні (залежно від стресів, різних захворювань), а за точністю тест не поступається вимірюванню глюкози крові. Аргумент проти — тест недостатньо стандартизований, і референсні межі істотно відрізняються в різних лабораторіях, більш висока вартість визначення; менша доступність визначення в деяких регіонах; неповна кореляція між рівнем HbA1c і середнім рівнем глюкози в деяких людей. Крім того, результати можуть бути хибними: при станах, що супроводжуються укороченням життя еритроцитів (гемолітична анемія, гіперспленізм, спадкові гемоглобінопатії, серпоподібноклітинна анемія, таласемія), крововтраті (як гострій, так і хронічній) спостерігається значне зниження HbAlc. У той же час його хімічна модифікація (ацетилювання при вживанні високих доз ацетилсаліцилової кислоти, ниркова недостатність), гіперліпідемія, гіпербілірубінемія призводять до завищення показників. У недалекому майбутньому процес стандартизації визначення HbAlc охопить більшість держав, що, ймовірно, дозволить раціоналізувати процес установлення діагнозу ЦД 2-го типу, а отже дасть можливість швидше діагностувати захворювання, таке небезпечне своїми віддаленими наслідками.
Численні дослідження показали, що частотні розподіли HbAlc мають однакові характеристики з глікемією плазми натще і постпрандіальною глікемією. Дослідженнями встановлено рівні HbA1c, за яких різко зростає вірогідність розвитку макро- або мікросудинних ускладнень. Крім того, HbA1c давно став одним з основних критеріїв ефективності лікування ЦД.
У 2010 р. Американська діабетична асоціація (ADA) запропонувала використовувати для діагностики ЦД також рівень HbA1c. До цього ADA не рекомендувала цей тест через відсутність стандартизації методу визначення. Однак на сьогодні існують стандартизовані методи визначення HbAlc, що дозволило експертам ADA рекомендувати його для діагностики ЦД із пороговим значенням понад 6,5 %. Як і з іншими тестами для діагностики ЦД, діагноз має бути підтверджений повторним визначенням HbA1c для виключення лабораторних помилок. Можлива діагностика ЦД при одночасному одноразовому визначенні HbA1c і глюкози. Згідно з рекомендаціями ADA, HbA1c в діапазоні 5,7–6,4 % відповідає категорії підвищеного ризику розвитку ЦД. До цієї ж категорії належить і стан ПТГ.
У 2011 р. ВООЗ схвалила використання показника HbA1c > 6,5 % як діагностичного критерію ЦД. При цьому діагностичний тест має бути виконаний із використанням методу визначення HbA1c, сертифікованого відповідно до National Glycohemoglobin Standardization Program (NGSP) і стандартизованого відповідно до референсних значень, прийнятих у дослідженні Diabetes Control and Complications Trial (DCCT), тобто нормальним вважається рівень до 6 %. Як і раніше, в разі відсутності симптомів гострої метаболічної декомпенсації діагноз має бути встановлений на підставі двох цифр, що перебувають у діапазоні ЦД (наприклад, двічі HbAlc або одноразове визначення HbA1c та одноразове визначення рівня глюкози). Згідно з рекомендаціями ВООЗ, рівень HbA1c 6,0–6,4 % сам по собі не дозволяє встановлювати будь-які діагнози.
Методи визначення глюкози в крові
На сьогодні як у лабораторіях, так і як засіб самоконтролю найчастіше використовують ензиматичні методи (глюкозооксидазний). Вони дають швидкий і доволі точний результат. Редукційні методи (Сомоджі — Нельсона, залізоціанідний), а також ортотолуїдиновий метод поступово виходять з практики. Стандартним методом для визначення глюкози відповідно до рекомендацій Міжнародної федерації клінічної хімії (IFCC) є визначення глюкози у венозній плазмі крові. Значення глюкози в плазмі приблизно на 11 % вище, ніж у цільній крові (при нормальному гематокриті). Венозні й капілярні зразки дають приблизно однакові значення рівня глюкози натще, але після їжі рівень глюкози в капілярній крові вищий. Значення глюкози в артеріальній крові приблизно на 7 % вище, ніж у венозній. Електрохімічні й фотометричні глюкометри мають доволі значний коефіцієнт відхилення від лабораторних значень (до 20 %), саме тому вони не повинні використовуватися для діагностики. На підставі таких вимірювань можна лише запідозрити наявність ЦД, остаточний діагноз необхідно встановлювати на підставі лабораторних методів дослідження глюкози.
Визначення рівнів С-пептиду та імунореактивного інсуліну
С-пептид — це білок, що відщеплюється від молекули проінсуліну в процесі синтезу інсуліну. Тому за кількістю С-пептиду можна умовно оцінити стан збереження інсуліносекреторної здатності бета-клітин підшлункової залози. Кількість циркулюючого С-пептиду еквівалентна кількості інсуліну. Дослідження С-пептиду проводять зазвичай для диференціальної діагностики ЦД 1-го і 2-го типів. При ЦД 1-го типу концентрація С-пептиду в крові низька або він відсутній взагалі, при ЦД 2-го типу рівень цього білка може тривалий час залишатися в межах нормальних значень або навіть бути підвищеним (гіперінсулінемія). Нормальний рівень С-пептиду при ЦД 2-го типу не може бути критерієм того, що хворий не потребує інсулінотерапії: вона призначається при незадовільному глікемічному контролі на тлі максимальної дози інших цукрознижувальних засобів незалежно від збереження інсуліносекреторної функції.
У багатьох випадках метод визначення імунореактивного інсуліну (ІРІ) можна використовувати для диференціальної діагностики ЦД 1-го і 2-го типів у хворих, які не отримували інсулінотерапію. Найчастіше його застосовують для діагностики інсуліном. Визначення ІРІ використовується для оцінки ступеня інсулінорезистентності й функціональної активності бета-клітин за індексами різних математичних моделей. Як і рівень С-пептиду, показник ІРІ не може бути критерієм для призначення інсулінотерапії при ЦД 2-го типу.
Обстеження на ЦД у групах ризику
ЦД 1-го типу належить до автоімунних захворювань, що характеризується наявністю автоантитіл до білкових структур на поверхні або всередині бета-клітин підшлункової залози. Наявність таких маркерів ще до розвитку явного захворювання може ідентифікувати стан ризику в пацієнта. Наприклад, особи з наявністю більше ніж одного виду автоантитіл (наприклад, ICA, IAA, GAD, IA-2) мають більший ризик. На сьогодні, однак, різні причини не дають змоги обстежувати людей із чинниками ризику для виявлення будь-яких автоімунних маркерів поза межами наукових клінічних досліджень. По-перше, граничні показники для деяких методів визначення імунних маркерів стосовно клінічного застосування достеменно не встановлені. По-друге, відсутній консенсус про подальші підходи в разі отримання позитивного результату тесту на автоантитіла. Отже, обстеження на автоантитіла може ідентифікувати осіб, у яких можна було б запобігти ЦД або відтермінувати його клінічний початок. На сьогодні економічна ефективність такого скринінгу залишається сумнівною принаймні до того часу, поки не буде доступною ефективна терапія.
У багатьох країнах в доволі значному відсотку випадків ЦД 2-го типу залишається недіагностованим. За оцінками експертів, кількість невиявленого ЦД 2-го типу навіть перевищує всі виявлені випадки. Хворі з невиявленим ЦД 2-го типу мають вірогідно підвищений ризик ІХС, інсульту та уражень периферичних артерій. Безумовно, раннє виявлення й подальше своєчасне лікування може значно зменшити тяжкість ЦД 2-го типу та його ускладнень.
Діагностика гестаційного цукрового діабету (ГЦД)
Діагностичні критерії ГЦД неодноразово переглядалися і змінювалися. Згідно з рекомендаціями ВООЗ, процедура скринінгу ГЦД ѓрунтується на ГТТ із 75 г глюкози і використанні аналогічних нормальних значень глікемії, що й для невагітних (табл. 2). При цьому до ГЦД відносять як ЦД, так і ПТГ (але не порушену глікемію натще), які виникли при вагітності.
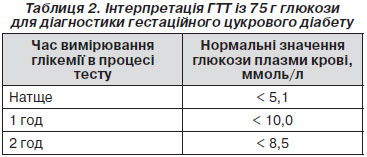
Критерії діагностики ГЦД: глікемія (в цільній капілярній крові) натще > 6,1 ммоль/л, через 2 год — > 7,8 ммоль/л або глікемія плазми венозної крові > 7,0 ммоль/л і через 2 год — > 7,8 ммоль/л.
Діагноз ГЦД встановлюється, якщо принаймні один із вказаних параметрів перевищує нормальні значення. В Україні критерії діагностики ГЦД відповідають критеріям ВООЗ.
Лабораторні особливості діагностики ЦД в осіб похилого віку
Діагностика ЦД в осіб похилого віку затруднена не лише через стерту клінічну картину захворювання, а й унаслідок нетипових особливостей лабораторної діагностики. До них відносяться: відсутність гіперглікемії натще в 60 % хворих; переважання ізольованої пост-прандіальної гіперглікемії в 50–70 % хворих; підвищення ниркового порогу для глюкози з віком.
Відсутність гіперглікемії натще і переважання гіперглікемії після їжі ще раз вказує, що в похилому віці при активному обстеженні для виявлення ЦД 2-го типу не слід обмежуватися епізодичними вимірюваннями рівня глюкози плазми (або капілярної крові) лише натще. Їх обов’язково необхідно доповнювати визначенням глікемії через 2 год після їжі.
У літньому віці при діагностиці ЦД або при оцінці його компенсації також не можна орієнтуватися лише на рівень глюкозурії. Якщо у молодих людей нирковий поріг для глюкози (тобто рівень глікемії, при якому глюкоза виникає в сечі) становить близько 10 ммоль/л, то після 65–70 років цей поріг зміщується до 12–13 ммоль/л. Отже, навіть незадовільна компенсація ЦД не завжди супроводжується появою глюкозурії.
Цукровий діабет 1-го типу: етіологія і патогенез
ЦД 1-го типу — імуноопосередкована або ідіопатична деструкція бета-клітин підшлункової залози, що призводить до абсолютної інсулінової недостатності. Це основний тип ЦД, що трапляється в дитячому й підлітковому віці. Частота первинної захворюваності на ЦД 1-го типу з віком знижується.
Для розвитку ЦД 1-го типу необхідна наявність генетичної схильності, що доведено наявністю різного ступеня асоціації із захворюванням деяких ділянок геному людини. У той же час для реалізації генетичної схильності необхідні чинники довкілля, які виступають як тригери автоімунного ураження бета-клітин підшлункової залози і сприяють виникненню клінічної картини хвороби. До таких чинників відносять різні віруси, низку інгредієнтів харчових продуктів, хімічні речовини.
Отже, на сьогодні ЦД 1-го типу можна розглядати як полігенне багатофакторне захворювання, що призводить до розвитку абсолютного інсулінового дефіциту, порушення вуглеводного, а потім й інших видів обміну речовин.
Співвідношення генетичних чинників і чинників довкілля може мати своє кількісне вираження у вигляді показника спадкування, що розраховується за методом Сміта. Його величина перебуває в прямій залежності від частоти повторних випадків захворювання в родинах хворих і у зворотній залежності від частоти захворювання в популяції. За даними різних авторів, коефіцієнт спадкування для всього ЦД 1-го типу, що виникає у віці від 0 до 40 років, у популяції становить 0,805, якщо прийняти повну залежність розвитку захворювання від генетичних чинників за 1. Це означає, що на 80 % розвиток ЦД 1-го типу залежить від спадкової схильності, а на 20 % — від чинників зовнішнього середовища.
Стадійність розвитку ЦД 1-го типу. Найбільш популярною теорією патогенезу ЦД 1-го типу впродовж останніх 25 років була теорія, запропонована в 1986 р. G. Eisenbarth. Згідно з його концепцією, в осіб з генетичною схильністю через певний час після впливу зовнішніх чинників індукується автоімунна реакція проти бета-клітин острівців Лангерганса, виникає клітинно-кероване руйнування бета-клітин, що характеризується появою клонів автореактивних Т-лімфоцитів із подальшим руйнуванням бета-клітин. Розгортається каскад біохімічних реакцій за участю цитокінів, макрофагів із синтезом окису азоту, вільних радикалів.
Згідно з цією теорією, руйнування бета-клітин можна схематично розділити на 6 стадій: 1 — генетична схильність; 2 — розвиток активного автоімунного процесу; 3 — зниження секреції інсуліну в першу фазу, що виявляється при проведенні внутрішньовенного глюкозотолерантного тесту; 4 — порушення толерантності до глюкози; 5 — клінічна маніфестація, що розвивається після загибелі 80–90 % бета-клітин із збереженою залишковою секрецією інсуліну; 6 — повна деструкція бета-клітин. Початкові стадії загибелі острівцевих клітин перебігають безсимптомно, але можуть бути виявлені за допомогою визначення автоантитіл. Лише на останніх стадіях процесу, коли переважна більшість бета-клітин загинула й виникла абсолютна недостатність інсуліну, наявні клінічні ознаки ЦД. Період від початку автоімунної агресії до розвитку клінічної картини ЦД 1-го типу може займати від декількох місяців (наприклад, у маленьких дітей) до 10 років і більше в дорослих пацієнтів.
За останні роки накопичилося досить багато нових даних про еволюцію патогенетичних змін бета-клітин острівців підшлункової залози при розвитку ЦД 1-го типу, у зв’язку з чим відома модель патогенезу ЦД 1-го типу G. Eisenbarth зазнала декількох уточнень (рис. 1) (Atkinson М.А., 2005).
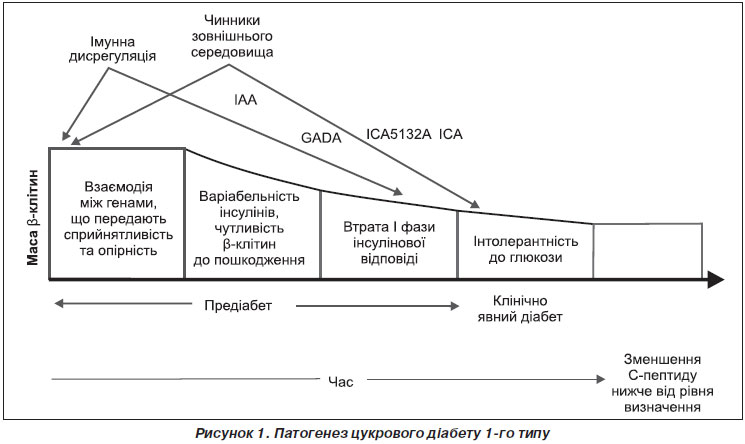
У ній розглядається взаємодія між генотипами, що визначають схильність до ЦД, і протективними генотипами, а не просто генетична схильність до ЦД. При цьому ці генотипи фактично впливають на сприйнятливість та опірність до ЦД 1-го типу впродовж усього періоду, що передує захворюванню на ЦД, а не лише перед автоімунною індукцією. На подальшій стадії розвивається інсуліт, унаслідок якого відбувається автоімунне руйнування бета-клітин із появою Т-активованих лімфоцитів, цитокінів і специфічних антитіл. Перебіг автоімунного процесу хвилеподібний з періодами затихання і новими загостреннями. Надалі відбувається втрата першої фази інсулінової секреції, потім — ПТГ і, зрештою, маніфестний ЦД 1-го типу.
Стадії загибелі бета-клітин в розвитку ЦД 1-го типу (за Atkinson М., 2005). При ЦД 1-го типу імунна система втрачає здатність нормально сприймати власні бета-клітини. Унаслідок цього відбувається автоімунне руйнування бета-клітин з активацією клітинної і гуморальної ланки імунітету, виникають плазматичні клітини, що секретують автоантитіла до різних антигенів бета-клітин, розвивається інсуліт — клітинно-керована автоімунна реакція проти бета-клітин із мононуклеарною інфільтрацією острівців.
У 1965 р. бельгійський учений W. Gepts першим описав лімфоцитарну інфільтрацію острівцевих клітин — інсуліт. За його даними, інсуліт спостерігався в 68 % випадків вперше виявленого ЦД 1-го типу. Через 20 років, у 1986 р., A. Foulis продемонстрував варіабельність інсулітів за матеріалами біопсії з різних госпіталів Великобританії з 1960 по 1985 рік. До сьогодні його колекція зразків вважається найвагомішою у світі. За його даними, інсуліт спостерігається в 78 % випадків вперше виявленого ЦД 1-го типу. У 2001 р. японським дослідником A. Imagawa і співавт. була виконана біопсія підшлункових залоз для виявлення ознак автоімунного процесу in vivo. Явища інсуліту й підвищена експресія антигенів були виявлені лише в 60 % пацієнтів, у решти 40 % жодних змін виявлено не було.
Лауреат премії Бантінга G. Eisenbarthу у 2010 р. навів фотографію зрізу підшлункової залози померлого пацієнта, який тривалий час хворів на ЦД 1-го типу. Була виявлена гетерогенна картина з лобулярними ділянками, де всі острівці містять бета-клітини, і, навпаки, лобулярні ділянки з острівцями без бета-клітин. Така гетерогенність, імовірно, пояснює повільне прогресування ЦД 1-го типу й значну частоту латентного автоімунного діабету дорослих з ознаками як ЦД 1-го типу, так і ЦД 2-го типу. У той же час подібний матеріал автопсії свідчить про відсутність інформативності проведення біопсії підшлункової залози.
Механізми взаємодії зовнішніх тригерних чинників та імунної системи організму. Упродовж останніх десятиліть вивчена велика кількість чинників довкілля, які вірогідно впливають на виникнення ЦД 1-го типу. Важлива роль відводиться природному компоненту. Так, між різними регіонами світу спостерігається величезна різниця в частоті захворюваності на ЦД 1-го типу. У сучасній моделі патогенезу ЦД 1-го типу розглядається не просто конкретна природна подія, що провокує захворювання, а природні пускові механізми й регулятори, відповідальні за розвиток ЦД. Вони діють протягом усього періоду, що передує самому захворюванню на ЦД.
Основні механізми дії тригерних чинників:
— активація поліклональних лімфоцитів (наприклад, інфекційними агентами);
— молекулярна мімікрія — ідентичність ділянок білкових послідовностей інфекційного чи хімічного агента й автоантигенів;
— підвищена імуногенність, що індукує імунну відповідь.
Ці механізми зрештою запускають розвиток автоімунних процесів, а також призводять до продукції різних автоантитіл, найбільш значущими з яких є автоантитіла до інсуліну (IAA), глутаматдекарбоксилази (GADA), тирозинфосфатазаподібного білка (IA-2A), транспортера цинку — автоантитіла до ZnT8, до острівцевих клітин (ICA).
На думку дослідників, тригерами можуть бути як інфекційні, так і неінфекційні чинники.
Інфекційні:
— ентеровіруси;
— ретровіруси;
— природжена краснуха;
— паразити;
— бактерії;
— грибки.
Неінфекційні:
— складові харчування:
- глютен, соя, інші рослини;
- коров’яче молоко, інсулін, глюкоза;
- ненасичені жири, антиоксиданти;
— тяжкі метали, нітрити/нітрати;
— токсини бета-клітин (ліки);
— психосоціальні чинники (стрес);
— ультрафіолетова радіація, температура/сезонність.
Віруси розглядаються як один з етіологічних чинників, що беруть участь у патогенезі ЦД 1-го типу. Епідеміологічні дослідження показали, що у хворих із вперше виявленими ЦД 1-го типу визначаються титри антитіл до специфічного для вірусу IGM, що свідчить на користь можливої ролі вірусу як тригера захворювання. З розвитком ЦД 1-го типу можуть бути асоційовані різні віруси — вірус Коксакі В, краснухи, епідемічного паротиту, цитомегаловірус, Епштейна — Барр, вітряної віспи тощо. Патогенетична роль вірусної інфекції в розвитку ЦД 1-го типу знаходить підтвердження в експериментальних дослідженнях A. Elshebani і співавт., які встановили, що штами ентеровірусу, отримані від хворих із вперше виявленим ЦД 1-го типу, можуть проникати в острівцеві клітки людини та індукувати їх деструкцію in vitro. На тваринних моделях показано, що розвиток діабету у мишей опосередкований вірусом енцефаломіокардиту М.
Віруси можуть втягуватися у патогенез ЦД 1-го типу принаймні двома різними шляхами: індукуючи автоімунітет проти бета-клітин або справляючи прямий ушкоджуючий вплив на бета-клітини.
Феномен антигенної мімікрії розглядається у випадку споживання з їжею новонародженими в перші 6 місяців життя сумішей на основі коров’ячого молока. Бичачий сироватковий альбумін, що міститься в коров’ячому молоці, подібний до антигену острівцевих клітин ICA 69 і може сприяти запуску автоімунної реакції.
Указується на взаємозв’язок стресових чинників, психосоціальних подій і дебюту ЦД 1-го типу.
Провокувати розвиток ЦД 1-го типу можуть хімічні речовини, у тому числі медикаменти (кортикостероїди, індометацин, вінкристин, циметидин і деякі інші). Азобарвник алоксан, токсичний до бета-клітин, і антибіотик стрептозотоцин, що вибірково руйнує бета-клітини, використовуються в моделях ЦД 1-го типу на тваринах.
Ключовим моментом у запуску імунної відповіді є взаємодія Т-клітинного рецептора з головним комплексом гістосумісності і пов’язаним з ним антигеном. Сучасна модель активації Т-клітин припускає участь двох сигналів. Перший специфічний сигнал надходить у момент зв’язування Т-клітинного рецептора (антигенного рецептора Т-клітин) з комплексом, що складається з молекули головного комплексу гістосумісності і пептидного антигена, який знаходиться на поверхні клітини. Однак одного цього сигналу недостатньо для активації Т-клітини. Необхідний інший неспецифічний сигнал, що надходить після з’єднання іншого рецептора (CD28) з його лігандами В7-1 (CD80) і В7-2 (CD86), розташованими на поверхні клітини. Якщо надійшли обидва сигнали, спостерігається активація Т-клітин, секреція цитокінів і подальша проліферація Т-клітин.
Центральним механізмом загибелі бета-клітин підшлункової залози при ЦД 1-го типу вважається апо-птоз. Апоптоз — програмована клітинна загибель, енергетично залежний, генетично контрольований процес, що запускається специфічними сигналами. На відміну від некрозу апоптоз ніколи не супроводжується запальною реакцією. Для нього характерне стиснення клітини, формування округлих, оточених мембраною апоптотичних тілець, які швидко фагоцитуються оточуючими клітинами. У процесі апоптозу клітина зникає безслідно протягом 15–120 хвилин.
Існує декілька шляхів реалізації програми клітинної смерті: за участю рецепторів плазматичної мембрани (Fas); за участю мітохондріального цитохрому С.
До загибелі бета-клітин залучені як Fas-залежні, так і незалежний механізми.
Поверхневий клітинний рецептор, що позначається Fas (CD95), взаємодіючи з відповідним лігандом (FASL) — трансмембранним білком, активується і запускає програму смерті клітки, що каскадно здійснюється специфічними цистеїновими протеазами. Життєздатність клітин залежить від співвідношення активаторів (Вах-білки) та інгібіторів (білки родини Bel) апоптозу.
Механізми розвитку ЦД 1-го типу (активація імунної відповіді, процес апоптозу тощо) перебувають під контролем генетичних чинників, які або сприяють, або оберігають організм конкретного індивідуума від розвитку специфічних імунних реакцій у відповідь на дію зовнішніх чинників.
У типових випадках діагноз ЦД 1-го або 2-го типу не становить труднощів. Проте останніми роками в деяких вікових групах трапляються форми захворювання, які за клінічними ознаками складно піддаються класифікації. Так, у віці після 35 років разом із ЦД 2-го типу часто трапляється і ЦД 1-го типу, що характеризується повільним прогресуванням. Цей особливий тип ЦД був названий LADA (Latent autoimmune diabetes melitus in adults), або автоімунний діабет у дорослих (Autoimmune diabetes in adults). Згідно з різними даними, у країнах Європи кількість пацієнтів із LADA становить близько 10 % від загальної кількості хворих на ЦД.
Клінічна картина захворювання в пацієнтів із LADA типова для ЦД 2-го типу і протягом 1–3 років компенсація вуглеводного обміну досягається застосуванням дієти і пероральних цукрознижувальних препаратів (ПЦЗП). Згодом розвивається інсулінова залежність. У цих хворих часто виявляють генетичні та імунологічні маркери, притаманні для ЦД 1-го типу. Для LADA характерні такі ознаки:
— вік дебюту зазвичай понад 35 років;
— клінічна картина ЦД 2-го типу без ожиріння;
— на початку — задовільний метаболічний конт-роль дієтою/ПЦЗП;
— розвиток інсулінової залежності через 1–3 роки;
— наявність маркерів ЦД 1-го типу: низький рівень С-пептиду; наявність автоантитіл до а-клітини (ICA і/або GAD).
Важливість проблеми своєчасної діагностики LADA у дорослих полягає в тому, що схожа клінічна картина захворювання, що маскується під ЦД 2-го типу, призводить до помилкової діагностики і призначення пероральних цукрознижувальних препаратів. Крім цього, наявні певні особливості в розвитку хронічних ускладнень ЦД, особливо макросудинних. Установлено, що ці пацієнти становлять групу високого ризику прогресування ураження серця. Усе це підкреслює значення і необхідність проведення вивчення генетичних, імунологічних і метаболічних маркерів ЦД 1-го типу, що на сьогодні є досить інформативними і доступними, для правильної діагностики і своєчасного лікування захворювання.
Цукровий діабет 2-го типу: етіологія і патогенез
Ще 1999 року ВООЗ охарактеризувала ЦД 2-го типу як метаболічне захворювання, що розвивається внаслідок порушення секреції інсуліну або зниженої чутливості тканин до дії інсуліну (тобто інсулінорезистентності). Суперечки про те, що з цих двох механізмів первинне, тривають. У проведених дослідженнях установлено, що в більшості хворих на ЦД 2-го типу погіршення тканинної чутливості до інсуліну є первинним або успадкованим дефектом. Якщо бета-клітини не здатні підтримувати достатньо високий рівень секреції інсуліну, щоб здолати інсулінорезистентність, розвивається гіперглікемія. Така послідовність подій притаманна як для хворих з ожирінням, так і для пацієнтів із нормальною масою тіла. Однак у незначної частини хворих на ЦД 2-го типу (здебільшого з нормальною масою тіла) первинний дефект може виникати на рівні бета-клітин і маніфестувати у вигляді порушення секреції інсуліну. Інсулінорезистентність у таких хворих розвивається одночасно або згодом. Обидва дефекти (як інсулінорезистентність, так і зменшення секреції інсуліну) спостерігаються в новонароджених дітей із низькою масою тіла. Маса тіла при народженні обернено пропорційна ризику розвитку ЦД 2-го типу у дорослих. Але який би дефект (тобто зниження секреції інсуліну чи інсулінорезистентність) не ініціював розвиток ЦД 2-го типу, важливим є той факт, що для виникнення значущого порушення толерантності до глюкози необхідні обидва механізми.
Інсулінорезистентність — це недостатня біологічна відповідь клітин на інсулін при його достатній концентрації в крові. Периферична інсулінорезистентність проявляється в порушенні поглинання глюкози периферичними тканинами, передусім печінкою, м’язами й жировою тканиною.
Виділяють 3 можливі рівні розвитку інсулінорезистентності: пререцепторний, рецепторний, пострецепторний.
Пререцепторний рівень полягає в генетично детермінованій продукції зміненої малоактивної молекули інсуліну, а також неповній конверсії проінсуліну в інсулін, що призводить до надлишку малоактивного проінсуліну.
Рецепторний рівень:
1. Мутації гену інсулінового рецептора, що призводять:
— до зниження швидкості біосинтезу інсулінового рецептора;
— погіршення внутрішньоклітинного транспорту й посттрансляційного процесингу;
— дефектів зв’язування інсуліну;
— зниження активності рецепторної тирозинкінази;
— пришвидшення деградації інсулінового рецептора;
— зниження афінності рецепторів до інсуліну.
2. Недостатня кількість інсулінових рецепторів (генетично обумовлена або набута як компенсаторна реакція на гіперінсулінемію).
Пострецепторний рівень:
— зниження активності тирозинкінази b-субодиниці;
— зменшення числа глюкозних транспортерів (GLUT);
— зниження активності двох основних ферментів утилізації глюкози — піруватдегідрогенази і глікогенсинтетази.
Особливе значення ожиріння в розвитку інсулінорезистентності обумовлене:
а) гіпертрофією адипоцитів при розвитку абдомінального типу ожиріння, оскільки при цьому відбувається зміна конформації молекули інсулінового рецептора й порушується процес його зв’язування з інсуліном;
б) підвищенням концентрації вільних жирних кислот (ВЖК) у плазмі крові, оскільки вони запобігають зв’язуванню інсуліну з гепатоцитами, пригнічують його гальмівну дію на глюконеогенез, що призводить до розвитку феномену ліпотоксичності. Крім цього, ВЖК порушують секрецію інсуліну а-клітинами, оскільки інгібують активність піруватдегідрогенази із зниженням утворення АТФ — найважливішого стимулятора секреції інсуліну.
Біологічна дія інсуліну полягає в регуляції метаболічних реакцій (обмін вуглеводів, жирів і білків) і мітогенних процесів (процесів росту, диференціювання тканин, синтезу ДНК, транскрипції генів). Тому сучасне поняття інсулінорезистентності не зводиться до параметрів, що характеризують лише метаболізм вуглеводів, а включає також зміни метаболізму жирів, білків, функції клітин ендотелію, експресії генів тощо. Термін «інсулінорезистентність» не слід ототожнювати із синдромом інсулінорезистентності, або метаболічним синдромом, описаним G. Reaven. До складу цього синдрому входять порушення толерантності до глюкози, абдомінальне ожиріння, дисліпідемія, артеріальна гіпертензія, гіперурикемія, гіперкоагуляція, мікроальбумінурія й деякі інші системні порушення. Інсулінорезистентність трапляється і при інших патологічних або фізіологічних станах, які не входять до поняття «метаболічний синдром»: при полікістозі яєчників, хронічній нирковій недостатності, інфекціях, терапії глюкокортикоїдами, вагітності, старінні.
За даними епідеміологічних досліджень, інсулінорезистентність спостерігається в 10 % осіб без метаболічних порушень; 58 % осіб з АГ (АТ > 160/95 мм рт.ст.); 63 % осіб із гіперурикемією (сечова кислота сироватки > 416 мкмоль/л у чоловіків і понад 387 мкмоль/л у жінок); 84 % осіб із гіпертригліцеридемією (тригліцериди понад 2,85 ммоль/л); 88 % осіб із низьким рівнем холестерину ліпопротеїнів високої щільності (нижче від 0,9 ммоль/л у чоловіків і нижче від 1,0 ммоль/л у жінок); 66 % осіб із ПТГ; 84 % осіб з ЦД 2-го типу.
При поєднанні ЦД 2-го типу (або ПТГ) з дисліпідемією, гіперурикемією і АГ, тобто з основними компонентами МС, частота виявлення інсулінорезистеності досягає 95 %. Це свідчить про те, що провідним механізмом розвитку метаболічного синдрому є інсулінорезистентність.
Вважають, що феномен інсулінорезистентності має міцну генетичну основу. Згідно з гіпотезою про «ощадливий генотип», висунутою V. Neel 1962 року, інсулінорезистентність — це еволюційно закріплений механізм виживання за несприятливих умов, коли періоди достатку чергувалися з періодами голоду. Наявність інсулінорезистентності забезпечувала накопичення енергії у вигляді відкладень жиру, запасів якого вистачало на те, щоб пережити голод. У процесі природного відбору насамперед закріплювалися ті гени, які забезпечували інсулінорезистентність і накопичення енергії. Гіпотеза підтверджується в експерименті на мишах, які переживали тривалий період голодування. Виживали лише ті миші, в яких була генетично опосередкована інсулінорезистентність. За сучасних умов у країнах із високим рівнем життя збережені в генетичній пам’яті механізми інсулінорезистентності продовжують працювати на накопичення енергії, що призводить до розвитку абдомінального ожиріння, дисліпідемії, АГ і ЦД 2-го типу.
При ЦД 2-го типу найбільше клінічне значення має втрата чутливості до інсуліну м’язової, жирової й печінкової тканин. Інсулінорезистентність м’язової тканини виявляється в зниженні надходження глюкози з крові в міоцити та її засвоєння в м’язових клітинах. Інсулінорезистентність жирової тканини виявляється в резистентності до антиліполітичної дії інсуліну, що сприяє накопиченню ВЖК і гліцерину. ВЖК надходять в печінку, де стають основним джерелом формування атерогенних ліпопротеїдів дуже низької щільності. Інсулінорезистентність тканини печінки характеризується зниженням синтезу глікогену й активацією процесів розпаду глікогену до глюкози (глікогеноліз) і синтезу глюкози de novo з амінокислот, лактату, пірувату, гліцерину (глюконеогенез), унаслідок чого глюкоза з печінки надходить у кровотік. Ці процеси в печінці активуються внаслідок відсутності їх пригнічення інсуліном.
Уперше гіпотеза про роль інсулінорезистентності в патогенезі ЦД 2-го типу виникла понад 60 років тому. Himsworth і Kerr використовували термін «інсулін-нечутливість» (синонім інсулінорезистентності) для опису слабкого зниження глікемії у відповідь на введення екзогенного інсуліну в огрядних хворих на ЦД. Інсулінорезистентність периферичних тканин передує розвитку ЦД 2-го типу і може виявлятися в найближчих родичів хворих на ЦД 2-го типу, які не мають порушень вуглеводного обміну. Тривало існуюча інсулінорезистентність компенсується надлишковою продукцією інсуліну бета-клітинами підшлункової залози (гіперінсулінемією), що підтримує вуглеводний обмін у нормі. Гіперінсулінемія прирівнюється до маркерів інсулінорезистентності і вважається провісником розвитку ЦД 2-го типу. Згодом при підвищенні ступеня інсулінорезистентності бета-клітини не справляються із збільшеним навантаженням глюкозою, що призводить до поступового виснаження інсуліносекреторної здатності бета-клітин і клінічної маніфестації ЦД. Насамперед страждає функція швидкої секреції інсуліну у відповідь на харчове навантаження (тобто перша фаза секреції інсуліну), тоді як друга фаза (фаза базальної секреції інсуліну) залишається надлишковою. Розвиток гіперглікемії ще більше підсилює інсулінорезистентність периферичних тканин і пригнічує інсуліносекреторну функцію бета-клітин. Цей механізм отримав назву «глюкозотоксичність».
Найбільше клінічне значення має втрата чутливості до інсуліну м’язової, жирової й печінкової тканин. При цьому жирова, печінкова і м’язова тканини мають не-однакову чутливість до інсуліну. Так, наприклад, у нормі для пригнічення на 50 % ліполізу в жировій тканині потрібно не більше 10 мкОД/мл інсуліну, для 50% пригнічення продукції глюкози печінкою — уже близько 30 мкОД/мл інсуліну, а для збільшення на 50% захвату глюкози м’язовою тканиною дозу інсуліну необхідно збільшити до 100 мкОД/мл. За ЦД 2-го типу вказані значення зміщуються вправо, тобто в бік збільшення інсулінорезистентності.
Отже, жирова тканина в нормі і за ЦД 2-го типу має мінімальний ступінь інсулінорезистентності, тканина печінки — проміжний, а м’язова тканина — максимальний. Тому в процесі розвитку ЦД 2-го типу, при поступовому виснаженні секреторної функції бета-клітин і відносному зменшенні гіперінсулінемії, спочатку знижується функція захвату глюкози м’язовою тканиною, потім страждає глікогеносинтетична функція печінки і в останню чергу відбувається зниження ліполітичної функції жирової тканини.
Відомо, що серцево-судинна смертність у хворих на ЦД 2-го типу в 3–4 рази вища, ніж в осіб без метаболічних порушень. Припускають, що в основі пришвидшеного атерогенезу й високої летальності від ІХС у хворих на ЦД 2-го типу лежить інсулінорезистентність і супутня гіперінсулінемія. Наявні клінічні докази того, що гіперінсулінемія є незалежним чинником ризику розвитку ІХС в осіб без ЦД 2-го типу. Гіперінсулінемія здійснює вагомий внесок у розвиток і прогресування атеросклерозу як в осіб, схильних до розвитку ЦД, так і у хворих на ЦД 2-го типу.
Гіперінсулінемія розглядається як непрямий маркер інсулінорезистентності. Встановлено чітку пряму залежність між ступенем інсулінорезистентності і вираженістю абдомінального ожиріння, атерогенністю ліпідного спектра крові, активацією системи коагуляції, а також товщиною стінки сонної артерії як в осіб без ЦД, так і у хворих на ЦД 2-го типу.
Роботи в галузі молекулярної біології і генетики показали, що у хворих на ЦД 2-го типу наявні генетичні дефекти, відповідальні за передачу сигналу після з’єднання інсуліну зі своїм рецептором (пострецепторні дефекти). Генетична схильність до інсулінорезистентності може не реалізуватися і не проявитися клінічно (у вигляді метаболічного синдрому і ЦД 2-го типу) за відсутності відповідних чинників довкілля: надмірного калорійного харчування і низької фізичної активності. Ці чинники самі по собі сприяють збільшенню абдомінального ожиріння, накопиченню ВЖК і, отже, посиленню наявної інсулінорезистентності.
Найпростішим і найзручнішим для використання в клінічній практиці методом оцінки інсулінорезистеності є зміна концентрації інсуліну плазми крові натще. Гіперінсулінемія при нормоглікемії зазвичай свідчить про наявність інсулінорезистентності і є провісником розвитку ЦД 2-го типу. Складною є стандартизація цього методу, оскільки нормальні значення інсулінемії вкрай варіабельні. Запропоновані різні індекси для оцінки інсулінорезистентності, що розраховуються за співвідношенням концентрацій інсуліну і глюкози плазми. Одним із таких загальноприйнятих розрахункових індексів є індекс НОМА, розроблений D. Matthews: інсулін сироватки натще (мкОД/мл) а аглюкоза плазми натще (ммоль/л) / 22,5. Індекс НОМА понад 2,7 розцінюється як стан інсулінорезистентності. Чим вищий індекс НОМА, тим нижча чутливість до інсуліну і, отже, вища інсулінорезистентність.
Інсулінорезистентність тканин не завжди супроводжується розвитком ЦД 2-го типу, але є тим провокуючим чинником, який перевіряє на міцність функціональну здатність бета-клітин підшлункової залози.
Стимуляція секреції інсуліну в нормі здійснюється здебільшого за допомогою глюкози. Установлено, що глюкоза спричинює деполяризацію мембрани, що призводить до руху екстрацелюлярного кальцію через кальцієві канали в середину бета-клітини. Ще 1968 року встановлена роль АТФ-чутливих калієвих каналів у регуляції секреції інсуліну.
Установлено, що в стані спокою заряд мембрани бета-клітини дорівнює приблизно 80 мВ. Визначені ділянки рецептора, які зв’язуються з вільним АТФ (або АДФ), і ділянки зв’язування Mg-АДФ. Від співвідношення концентрації цих метаболітів залежить стан калієвих каналів. Деполяризація мембрани призводить до зниження заряду до 50 мВ і закриття калієвих каналів. Це сприяє відкриттю вольтажзалежних кальцієвих каналів і призводить до підвищення концентрації внутрішньоклітинного кальцію.
У нормі підшлункова залоза дорослої людини секретує 35–50 ОД інсуліну на добу, що становить 0,6–1,2 ОД на кг маси тіла на добу. Ця секреція поділяється на харчову (стимульовану) і базальну.
Базальна секреція інсуліну наявна за відсутності будь-яких екзогенних стимулів секреції інсуліну. Роль базальної секреції інсуліну полягає в зниженні базальної продукції глюкози печінкою, рівня глюкози натще, рівня ВЖК.
Базальна секреція інсуліну in vivo завжди визначається вранці після нічного голодування. Базальна секреція інсуліну забезпечує оптимальний рівень глікемії й анаболізму в інтервалах між їжею і під час сну. Базальний інсулін секретується із швидкістю приблизно 1 ОД/год, при тривалому фізичному навантаженні або тривалому голодуванні секреція зменшується до 0,5 ОД/год. На прандіальний інсулін припадає не менше 50–60 % від добової продукції інсуліну.
Харчова (стимульована) секреція інсуліну відповідає постпрандіальному підвищенню рівня глікемії, тобто за її рахунок забезпечується нейтралізація гіперглікемізуючої дії їжі. Кількість цього інсуліну приблизно відповідає кількості прийнятих вуглеводів — приблизно 1–1,5 ОД на 10–12 г вуглеводів. У результаті забезпечується оптимальний, насамперед для центральної нервової системи, рівень глікемії в межах 3,3–8,4 ммоль/л.
Перша фаза секреції інсуліну реєструється після внутрішньовенного введення глюкози й забезпечується швидким наростанням рівня іонів кальцію в бета-клітині. У бета-клітині виявлено два пули інсулінових гранул, кожен з яких має своє особливе значення. Так, один із них створює ранній пік секреції інсуліну шляхом негайної інсулінової відповіді. Цей пул гранул найбільш лабільний. Інша фаза секреції інсуліну, більш тривала за часом, забезпечується стабільним пулом гранул.
У людини ранній пік секреції інсуліну виявляється в процесі проведення внутрішньовенного глюкозотолерантного тесту. Ранній пік секреції інсуліну спричинює швидке пригнічення продукції глюкози печінкою, контролюючи збільшення глікемії; пригнічує ліполіз і секрецію глюкагону; підвищує чутливість периферичних тканин до дії інсуліну, сприяючи засвоєнню глюкози; обмежує рівень прандіальної глікемії.
При розвитку ЦД ранній пік секреції інсуліну відсутній.
Крім того, при використанні препаратів, що блокують вольтажзалежні кальцієві канали або активують АТФ-залежні калієві канали, також відбувається зниження ранньої фази секреції інсуліну. Так, до порушень секреції інсуліну при ЦД 2-го типу відносять: зниження секреції інсуліну у відповідь на глюкозу та інші стимулятори; порушення пульсаторної секреції інсуліну; порушення перетворення проінсуліну в інсулін, що призводить до підвищення вмісту проінсуліну.
Секреція інсуліну може зменшуватися внаслідок порушення внутрішньоутробного розвитку підшлункової залози через недостатнє живлення плода і в пост-натальному періоді, вплив глюкозотоксичності (посилює дефекти секреції інсуліну), генетичні дефекти в механізмах секреції (мутації генів інсуліну, глюкокінази, транспортера глюкози GLUT-2 тощо).
Як відомо, кількість бета-клітин визначає можливості вироблення інсуліну підшлунковою залозою. Зниження маси бета-клітин може становити 20–40 % у хворих на ЦД 2-го типу. При цьому морфологічно острівці Лангерганса залишаються нормальними, без ознак інсуліту. Етіологія зменшення маси бета-клітин залишається не до кінця з’ясованою. З віком кількість функціонуючих бета-клітин не зменшується. Ожиріння, інший інсулінорезистентний стан супроводжуються підвищенням маси бета-клітин. При цьому для розвитку інсулінопенії й потім явного ЦД необхідне зниження маси бета-клітин на 80–90 %. Більше того, до кінця ще невідомо, чи є на ранніх стадіях розвитку ЦД 2-го типу будь-яке зниження бета-клітинної маси. Очевидно, існують й інші чинники, крім зменшення маси бета-клітин, відповідальні за погіршення секреції інсуліну. Вони можуть полягати у втраті чутливості або відсутності реакції бета-клітин на збільшення концентрації глюкози в крові, результатом чого є втрата нормальної секреторної інсулінової відповіді на гіперглікемію.
Звертають на себе увагу публікації про можливий вплив аміліну на дефект у секреції інсуліну. Цей пептид, що складається з 37 амінокислот, секретується бета-клітинами разом з інсуліном. Було показано, що він є попередником для утворення депозитів амілоїду, які часто спостерігаються у хворих на ЦД 2-го типу. Як було встановлено на тваринних моделях, амілін у великих дозах інгібує секрецію інсуліну. Після своєї секреції амілін накопичується позаклітинно, і ці депозити можуть призвести до дисфункції бета-клітин, погіршуючи транспорт з крові стимуляторів секреції інсуліну або впливаючи на глюкозочутливий і/або інсуліносекретуючий апарат бета-клітини.
Переконливі дані про зниження функції бета-клітин були отримані при проведенні Британського проспективного дослідження цукрового діабету (UKPDS). За даними UKPDS, коли у хворих уперше розвивалася симптоматика ЦД 2-го типу, у них вже було втрачено приблизно 50 % функції бета-клітин. Група дослідників UKPDS аналізувала дані про вплив дієти і терапії препаратами сульфонілсечовини у хворих на ЦД 2-го типу без ожиріння і вплив дієти, терапії препаратами сульфонілсечовини і метформіном у хворих на ЦД 2-го типу в поєднанні з ожирінням на функцію бета-клітин упродовж перших 6 місяців терапії. Препарати сульфонілсечовини спочатку посилювали функцію бета-клітин, однак згодом відзначалося прогресуюче зниження їх функції, розпочинаючи з першого року терапії. Функція бета-клітин прогресивно погіршувалася також і при терапії метформіном. Дані дослідження показали, що при ЦД 2-го типу відзначається прогресуюче зниження функції бета-клітин, що розпочинається за багато років до розвитку клінічної симптоматики захворювання і невпинно прогресує. На цей процес не впливають ні дієтотерапія, ні терапія препаратами сульфонілсечовини або метформіном.
Механізм, відповідальний за прогресуюче зниження функції бета-клітин, до кінця не з’ясований. Окремі дослідження вказують, що збільшення частоти апоптозу і зниження регенерації бета-клітин генетично запрограмовані, тобто є наслідком генетичних порушень. Надмірна секреція інсуліну в ранній період інсулінорезистентності може призводити до збільшення апоптозу клітин, а супутня надлишкова секреція аміліну сприяє розвитку амілоїдозу острівців.
Отже, в основі розвитку ЦД 2-го типу лежить виражена інсулінорезистентність, але за відсутності дефекту секреції інсуліну вона сама по собі ніколи не призведе до розвитку ЦД. Тому функціональна неповноцінність бета-клітин підшлункової залози є пусковим механізмом у розвитку ЦД 2-го типу.
Усі механізми формування ЦД 2-го типу (інсулінорезистентність, дефект секреції інсуліну, знижений інкретиновий ефект, дефект секреції глюкагону, гіпер-продукція глюкози печінкою) неминуче завершуються розвитком вираженої гіперглікемії. Тривале існування гіперглікемії токсичне не лише щодо периферичних органів і тканин, а й щодо бета-клітин, що продукують інсулін. Гіперглікемія знижує здатність бета-клітин секретувати адекватні кількості інсуліну у відповідь на введення глюкози і може також певною мірою збільшувати інсулінорезистентність. Цей феномен має назву «глюкозотоксичність». Хронічне збільшення рівня глікемії пов’язане з подальшим посиленням інсулінорезистентності й печінкової продукції глюкози з подальшим погіршенням здатності бета-клітин секретувати інсулін. Отже, гіперглікемія є не лише наслідком, але й причиною подальшого погіршення глюкозотолерантності у хворого на ЦД. Зниження рівня глікемії будь-яким способом (режим харчування, фізична активність, використання препаратів, що стимулюють секрецію інсуліну, самого інсуліну) призводить до поліпшення інсуліночутливості і секреції інсуліну, зниження продукції глюкози печінкою. Погіршення роботи бета-клітин функціональне за своєю природою і не може бути спричинене їх загибеллю. Інакше не спостерігалося б відновлення функції бета-клітин при поліпшенні глікемічного контролю. Запропоновано багато механізмів для того, щоб пояснити, як гіперглікемія знижує секрецію інсуліну. Так, один із механізмів полягає в тому, що збільшення внутрішньоклітинної кількості похідних глюкозаміну інгібує інсулінозалежний транспорт глюкози в бета-клітину. Інший механізм обумовлений тим, що зниження рівня внутрішньоклітинного малоніл-КоА призводить до збільшення окиснення ВЖК. Отже, на ранніх стадіях розвитку ЦД 2-го типу секреція інсуліну може і не відрізнятися від аналогічного показника у здорових осіб такого ж віку і маси тіла. Вважається, що первинним порушенням при ЦД 2-го типу і ПТГ є зниження чутливості периферичних тканин до інсуліну, а погіршення роботи бета-клітин виникає вторинно як механізм, скерований на компенсацію інсулінорезистентності.
Хронічна гіперглікемія і глюкозотоксичність — основні інструменти прогресування ЦД 2-го типу і втрати інсуліносекретуючої функції бета-клітин підшлункової залози.
У розвитку і прогресуванні ЦД 2-го типу можна виділити декілька етапів. Уперше припущення про поетапне згасання бета-клітин підшлункової залози при ЦД 2-го типу висловив американський вчений Г. Вейр (G.C. Weier), який працює в Джослінівськом центрі (Бостон, США). Згідно з його гіпотезою, виділяють 5 етапів втрати маси і функції бета-клітин при ЦД (табл. 3).
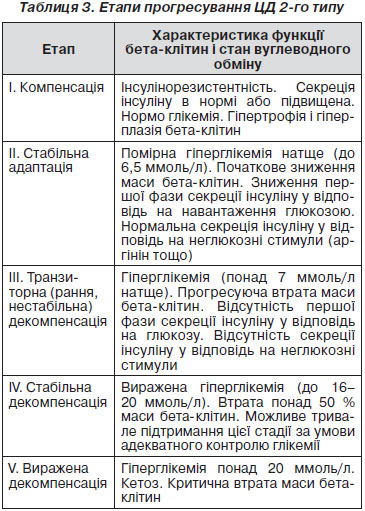
Згідно з G. Weier, при ЦД 2-го типу можливий перехід не лише від I стадії до IV, але й у зворотному напрямку (від IV до II або навіть I стадії). Це можливо суто гіпотетично при своєчасному початку лікування ЦД 2-го типу і при застосуванні препаратів, які сприяють збереженню і відновленню функціонуючої маси бета-клітин.
Основні патофізіологічні аспекти ЦД 2-го типу в похилому віці
Відомо, що старіння організму супроводжується фізіологічною перебудовою і не обминає жодного органа. З віком неминуче виникають проблеми в роботі серця, легенів, нервової системи. Починаючи з 50–60 років у частини людей відбувається необоротний процес зниження толерантності до глюкози, що розглядається як основний патогенетичний механізм розвитку ЦД 2-го типу.
Вікові зміни толерантності до глюкози характеризуються такими тенденціями. Після 50 років за кожні подальші 10 років глікемія натще збільшується на 0,055 ммоль/л, а глікемія через 2 год після їжі збільшується на 0,5 ммоль/л.
Як бачимо, найбільших змін зазнає постпрандіальна глікемія, тоді як глікемія натще змінюється незначно. Що ж лежить в основі вікових порушень толерантності до глюкози?
Для відповіді на це питання необхідно прослідкувати, як змінюються з віком основні механізми, що відповідають за метаболізм глюкози: чутливість тканин до інсуліну; секреція інсуліну підшлунковою залозою у відповідь на харчове навантаження; продукція глюкози печінкою.
В осіб похилого віку за допомогою гіперглікемічного клемпу виявлено зниження чутливості периферичних тканин до інсуліну і, відповідно, зниження захвату глюкози периферичними тканинами. Цей дефект здебільшого виявляється в осіб із надмірною масою тіла. Дослідження з використанням молекулярно-біологічних технологій показали, що інсулінорезистентність в похилому віці не пов’язана з патологією рецепторів до інсуліну. У той же час з віком виявлено чітке зниження активності тирозинкінази рецепторів інсуліну в м’язовій тканині. На сьогодні немає чіткого розуміння, чи інсулінорезистентність є звичайним процесом старіння організму, чи вона виникає внаслідок зміни способу життя в старшому віці, тобто зміни характеру харчування, зниження фізичної активності, розвитку абдомінального ожиріння.
Безумовно, люди старшого віку через різні причини (у тому числі соціально-економічні) віддають перевагу дешевій калорійній їжі, що містить надлишок насичених жирів і легкозасвоюваних цукрів. Багато людей мають супутні захворювання і приймають низку препаратів, які можуть негативно впливати на вуглеводний обмін (наприклад, тіазидні діуретики, неселективні бета-блокатори, стероїдні препарати, психотропні засоби тощо). Нерідко супутні захворювання (патологія серця, легенів, опорно-рухового апарату тощо) обмежують фізичну активність літніх людей. Тому з віком зростає число осіб з ознаками метаболічного синдрому. Частота ожиріння збільшується до 70-річного віку, а потім зазвичай знижується. Для опису літніх людей іноді використовують термін «саркопенічне ожиріння», що характеризує осіб із надмірною масою тіла, порушенням толерантності до глюкози або ЦД, які мають істотне зниження м’язової сили і м’язової маси.
Механізм розвитку саркопенічного ожиріння пов’язаний із заміщенням м’язової тканини жировими клітинами. Зниження м’язової маси — одна з вагомих причин розвитку інсулінорезистентності в осіб похилого віку, оскільки м’язова тканина належить до тих периферичних тканин, які в нормі захвачують глюкозу з кровотоку, тим самим знижуючи глікемію.
До причин зниження м’язової маси в осіб похилого віку належать низька фізична активність і детренованість; гормональний дисбаланс (зниження активності ендогенних статевих стероїдів, що забезпечують анаболічні процеси в організмі); активація субклінічного запалення, що виснажує і руйнує м’язові волокна; процеси глікозилювання білків, що входять до складу м’язових волокон; зниження запасу вітамінів і розвиток анемії (зокрема, дефіцит еритропоетину).
В осіб похилого віку без надмірної маси тіла встановлено істотне зниження першої фази секреції інсуліну. Можливо, саме з цим пов’язане виражене підвищення постпрандіальної глікемії (на 0,5 ммоль/л) кожне десятиліття після 50-річного віку. Молекулярно-біологічні дослідження показали, що у літніх осіб із нормальною масою тіла знижена активність гена глюкокінази, що забезпечує чутливість бета-клітин підшлункової залози до стимулюючої дії глюкози. Дефект цього гена може пояснити недостатню секрецію інсуліну у відповідь на введення глюкози.
Зміни метаболізму глюкози в печінці не можуть лежати в основі виражених вікових змін толерантності до глюкози. Непрямим свідченням нормальної продукції глюкози печінкою в осіб похилого віку є той факт, що глікемія натще (багато в чому залежить від викиду глюкози печінкою в нічний час) з віком змінюється не-істотно.
Отже, в осіб похилого віку основними механізмами, що визначають вікове порушення толерантності до вуглеводів і розвиток ЦД, є посилення інсулінорезистентності периферичних тканин і зниження секреції інсуліну (особливо ранньої фази) у відповідь на харчове навантаження.

1. Генделека Г.Ф. Превентивная диабетология. — Одесса: ВМВ, 2013. — 608 с.
2. Дедов И.И. Сахарный диабет — опаснейший вызов мировому сообществу / И.И. Дедов // Вестник РАМН. — 2012. — № 1. — С. 7-13.
3. Древаль А.В. Распространенность сахарного диабета 2 типа и других нарушений углеводного обмена в зависимости от критериев диагностики / А.В. Древаль, И.В. Мисникова, И.А. Барсуков [и др.] // Сахарный диабет. — 2010. — № 4. — C. 116-121.
4. Ендокринологія. Підручник для студентів вищих мед. навч. закладів / За ред. проф. П.М. Боднара. — Вінниця: Нова Книга, 2010. — 464 с.
5. Кравчун Н.А., Казаков А.В., Караченцев Ю.И. и др. Сахарный диабет 2 типа: скрининг и факторы риска. — Х.: Новое слово, 2010. — 256 с.
6. Маньковський Б.М. Показники компенсації ЦД в Україні — результати дослідження «Діаконтроль» [Текст] / Б.М. Маньковський, О.С. Ларін, Л.В. Бертаєва // Клінічна ендокринологія та ендокринна хірургія. — 2007. — № 4. — С. 46-48.
7. Сахарный диабет: диагностика, лечение, профилактика / Под ред. И.И. Дедова, М.В. Шестаковой. — М.: Медицинское информационное агентство, 2011. — 808 с.
8. Тронько М.Д. Гендерні та статеві особливості цукрового діабету. [Текст] / М.Д. Тронько. — К.: РИА Триумф, 2008. — 208 с.
9. Cowie C.C. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health And Nutrition Examination Survey 1999-2002 / C.C. Cowie, K.F. Rust, D.D. Byrd-Holt [et al.] // Diabetes Care. — 2006. — Vol. 29. — P. 1263-1268.
10. Dall T.M., Zhang Y., Chen Y.J. et al. The economic burden of diabetes // Health Aff. — 2010. — Vol. 29, № 2. — P. 297-303.
11. Gregg E.W. Trends in the prevalence and ratio of diagnosed to undiagnosed diabetes according to obesity levels in the U.S. / E.W. Gregg, B.L. Cadwell, Y.J. Cheng [et al.] // Diabetes Care. — 2004. — Vol. 27. — P. 2806-2812.
12. IDF Diabetes Atlas. Sixth edition. — International Diabetes Federation, 2013. http://www.idf.org / diabetesatlas
13. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus [Text] // Diabetes Care. — 2002. — Vol. 25, Suppl. 1. — P. 5-20.
14. Standards of medical care in diabetes — 2013. American Diabetes Association // Diabetes Care. — 2013. — Vol. 36 (Suppl. 1). — S. 11-S.66.
15. Wild S., Roglis G., Green A. et al. Global prevalence of diabetes: estimates for the year 2000 and projection for 2030 / S. Wild, G. Roglis, A. Green [et al.] // Diabetes Care. — 2004. — Vol. 27, № 5. — P. 1047-1053.