
Международный эндокринологический журнал 3 (59) 2014
Вернуться к номеру
Гликемическая память как патогенетическое основание для формирования алгоритма современной антидиабетической терапии
Авторы: Полторак В.В., Красова Н.С. - ГУ «Институт проблем эндокринной патологии им. В.Я. Данилевского НАМН Украины», г. Харьков; Горшунская М.Ю. - Харьковская медицинская академия последипломного образования
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
Предупреждение/замедление развития сосудистых осложнений остается одной из главных задач в лечении сахарного диабета. Эпидемиологические исследования показали недостаточную эффективность жесткого гликемического контроля у пациентов с длительно существующим диабетом. Данный феномен, подтвержденный на животных моделях и исследуемый на молекулярно-генетическом уровне, называют метаболической/гликемической памятью и связывают с эпигенетическими модификациями генной экспрессии. С другой стороны, доказано, что раннее интенсивное вмешательство при сахарном диабете 1-го и 2-го типа снижает риск развития и прогрессирования микро- и макрососудистых осложнений, формируя основу для отдаленных благоприятных эффектов, которые сохраняются за пределами нормогликемии. Вышеизложенное обосновывает изменение тактики лечения сахарного диабета с момента постановки диагноза для раннего и максимально безопасного достижения уровней гликемии и гликозилированного гемоглобина, близких к нормальным.
Попередження/уповільнення розвитку судинних ускладнень залишається одним із головних завдань у лікуваннi цукрового діабету. Епідеміологічні дослідження довели недостатню ефективність жорсткого глікемічного контролю в пацієнтів із тривало існуючим діабетом. Цей феномен, підтверджений на тваринних моделях і досліджуваний на молекулярно-генетичному рівні, називають метаболічною/глікемічною пам’яттю і пов’язують з епігенетичними модифікаціями генної експресії. З іншого боку, доведено, що раннє інтенсивне втручання при цукровому діабеті 1-го та 2-го типу знижує ризик розвитку та прогресування мікро- і макросудинних ускладнень, формуючи підґрунтя для віддалених сприятливих ефектів, які зберігаються за межами нормоглікемії. Вищевикладене обґрунтовує зміну тактики лікування цукрового діабету з моменту встановлення діагнозу для раннього й максимально безпечного досягнення рівнів глікемії та глікозильованого гемоглобину, близьких до нормальних.
Prevention/delay of the development of vascular complications remains one of major challenges in treatment of diabetes mellitus. Epidemiological studies have shown lack of efficacy of stable glycemic control in patients with long-existing diabetes. This phenomenon, confirmed in animal models and analyzed at the molecular genetic level, is called metabolic/glycemic memory and associated with epigenetic modifications of gene expression. On the other hand, it has been proven that early intensive intervention in type 1 and 2 diabetes mellitus reduces the risk of micro- and macrovascular complications development and progression, forming the basis for long-term favorable effects that persist beyond normoglycemia. The foregoing justifies change of therapeutic approach in diabetes mellitus since the moment of establishing diagnosis for the early and maximum safely achievement of blood glucose and glycosylated hemoglobin levels close to normal ones.
гликемическая память, эпигенетика, сахарный диабет, сосудистые осложнения.
глікемічна пам’ять, епігенетика, цукровий діабет, судинні ускладнення.
glycemic memory, epigenetics, diabetes mellitus, vascular complications.
Статья опубликована на с. 15-21
Сосудистые осложнения остаются главной причиной развития инвалидизирующих нарушений и повышенной смертности при сахарном диабете (СД) как 1-го, так и 2-го типа. Хотя большинство оригинальных клинических исследований диабетической популяции часто фокусируется на микрососудистых конечных точках, таких как ретинопатия и нефропатия, возрастающее внимание направлено на макрососудистые осложнения, клинически представленные инфарктом миокарда, инсультами и заболеванием периферических сосудов.
Необходимость контроля гликемии, т.е. достижения целевых значений, максимально приближенных к физиологическим показателям, для торможения развития и прогрессирования в первую очередь сердечно-сосудистых осложнений при СД 1-го и 2-го типа была доказана в многочисленных эпидемиологических и клинических исследованиях. Так, крупнейший анализ индивидуальных данных по 104 проспективным испытаниям с общим количеством участников свыше 1,1 млн человек, The Emerging Risk Factors Collaboration, продемонстрировал, что повышенные уровни глюкозы даже в недиабетическом диапазоне связаны со значительным увеличением как общего риска смертности, так и риска смертности от рака и сосудистых заболеваний [1]. В ставшем классическим исследовании DCCT (Diabetes Control and Complication Study) с последующим многолетним (1994–2006) наблюдением его участников (1441 пациент) в испытании EDIC (Epidemiology of Diabetes Intervention and Complications) было показано, что ранняя интенсивная инсулинотерапия лиц с СД 1-го типа существенно снизила не только уровень гликозилированного гемоглобина в период активного лечения, но и риск развития отдаленных, оцененных в течение последующих 10 лет наблюдений, микро- (ретинопатия, периферическая и сердечная автономная нейропатия и нефропатия) и макрососудистых осложнений [2–6]. Необходимо отметить, что после окончания активной фазы исследования и перевода всех пациентов на интенсивную терапию во всех группах наблюдался сопоставимый, близкий к нормальному уровень гликозилированного гемоглобина.
Корреляция между ранним гликемическим контролем и сердечно-сосудистым прогнозом отмечается и у больных СД 2-го типа, что было показано в исследовании UKPDS (United Kingdom Prospective Diabetes Study, 3867 пациентов), в том числе при анализе результатов обследования, проведенного через 10 лет после завершения основного испытания [7, 8]. Привлекает внимание тот факт, что на фоне потери различий в показателе гликозилированного гемоглобина между группами с интенсивной и традиционной терапией уже через 1 год после прекращения активной фазы испытания положительное влияние ранней интенсивной терапии как инсулином с сульфонилмочевиной, так и метформином сохранялось и через 10 лет. А именно: наблюдалось снижение относительного риска связанных с диабетом конечных точек (на 9 %, Р = 0,004, и 21 %, Р = 0,01, соответственно), микрососудистых заболеваний (на 24 %, Р = 0,001, и 16 %, Р = 0,31), а также существенное снижение риска инфаркта миокарда (на 15 %, Р = 0,01, и 33 %, Р = 0,005) и общей смертности (на 13 %, Р = 0,01, и 27 %, Р = 0,002).
Результаты пятилетнего исследования ADVANCE (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation), в которое было включено 11 140 пациентов с СД 2-го типа из 20 стран мира, также показали, что только в группе с длительностью СД менее 5 лет и исходным отсутствием атеросклеропатии снижение гликозилированного гемоглобина до 6,5 % и ниже достоверно (на 10 %, Р < 0,013) уменьшает риск развития сердечно-сосудистых осложнений [9]. Характерным является то, что данные исследования продемонстрировали наличие отдаленных положительных эффектов раннего интенсивного гликемического контроля при СД 1-го и 2-го типа. Именно с наличием длительно предсуществующей гипергликемии и ассоциированных сердечно-сосудистых факторов риска связывают отсутствие выраженного положительного влияния жесткого гликемического контроля на конечные сердечно-сосудистые точки в исследованиях VADT (Veterans Affairs Diabetes Trial) и ACCORD (Action to Control Cardiovascular Risk in Diabetes) [10, 11]. Полученные результаты позволили клиницистам «воскресить» сформулированный экспериментаторами еще в 1980-е годы феномен, описываемый терминами «метаболическая/гликемическая память» и «эффект наследия» (legacy effect).
На настоящий момент молекулярные механизмы, лежащие в основе данного феномена, установлены не полностью и активно изучаются. Сама концепция явилась результатом новаторского исследования Engerman и Kern [12], проведенного на собаках с аллоксановым диабетом. Было обнаружено, что у животных с плохим в течение 2,5 года гликемическим контролем его нормализация на такой же срок практически не уменьшала симптомы развития ретинопатии по сравнению с животными, имевшими плохой гликемический контроль в течение всего срока исследования (5 лет). В то же время интенсивная инсулинотерапия, обеспечившая нормальный гликемический контроль на протяжении 5 лет, в значительной степени тормозила развитие данного микрососудистого осложнения. Более поздние исследования на мышах и крысах со стрептозотоциновым или генетически обусловленным диабетом, а также на собаках с диабетом или галактоземией обнаружили сходный феномен не только относительно ретинопатии, но и в процессе развития нефропатии и атеросклероза [13–15]. Толчком к пониманию конкретных механизмов, формирующих метаболическую память, явились эксперименты на клеточных культурах, которые показали, что временное воздействие на эндотелиальные клетки высоких уровней глюкозы с последующей их нормализацией сопровождалось персистентной активацией белков внеклеточного матрикса, в том числе фибронектина и коллагена IV типа [16]. Для объяснения полученных результатов авторы постулировали роль эпигенетических механизмов регуляции биологических процессов.
Впервые термин «эпигенетика» был использован в отношении комплекса взаимодействий между геномом и окружающей средой, вовлеченного в процессы развития и дифференциации высших организмов. В настоящее время этот термин используется по отношению к наследуемым изменениям, которые не связаны с изменениями в последовательности ДНК.
Таким образом, эпигенетика в целом может быть охарактеризована как сумма механизмов, необходимых для реализации генетической программы развития организма. В частности, включение и выключение экспрессии конкретных генов обеспечивается такими установленными на настоящий момент биохимическими процессами, как метилирование ДНК по цитозину (так называемые долгосрочные эпигенетические модификации) и модификации гистоновых белков (так называемые краткосрочные модификации: ацетилирование/деацетилирование, фосфорилирование/дефосфорилирование, метилирование, убиквитинирование) [17]. Вышеперечисленные эпигенетические модификации изменяют доступность ДНК и структуру хроматина и представляют собой основу для регуляции процессов считывания генетической информации. Кроме того, в последнее время к эпигенетике относят альтернативный сплайсинг генных транскриптов (образование изоформ белка) и влияние на экспрессию некодирующих малых (микро) РНК, что также не затрагивает последовательность оснований в молекулах ДНК [18]. Важным является тот факт, что все эти регуляторные механизмы подвержены внешнему влиянию и в конечном итоге представляют собой результат суммы изменений под воздействием окружения, формируя конкретный фенотип или патофенотип [19].
Уникальное исследование, наглядно продемонстрировавшее возникновение в течение жизни эпигенетических модификаций под воздействием различных внешних факторов, было проведено с участием близнецов (80 обследуемых в возрасте от 3 до 74 лет) [20]. Данная работа позволила получить молекулярно-генетическое объяснение развития со временем фенотипических различий у имеющих одинаковый генотип (однояйцевых) близнецов. Известно, что большинство пар однояйцевых близнецов не являются идентичными, у них часто наблюдаются различия в чувствительности к заболеваниям и широкая антропометрическая вариабельность. Исследователи обнаружили, что у однояйцевых близнецов, эпигенетически идентичных в раннем периоде жизни, с возрастом отмечаются выраженные различия как общего содержания, так и распределения по геному метилированных участков ДНК и сайтов ацетилирования гистонов, что изменяет индивидуальный характер экспрессии генов. При этом различия были тем более выраженными, чем больше и дольше различались условия проживания близнецов из пары.
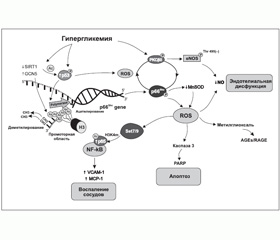
Одним из факторов, которые вызывают существенные эпигенетические изменения и активно изучаются в настоящее время, является гипергликемия. Доказано, что даже кратковременное повышение уровня глюкозы вызывает персистентные эпигенетические модификации и меняет экспрессию генов при последующей нормогликемии [21]. Целый ряд исследований in vitro и in vivo выявили конкретные биохимические процессы-«посредники», такие как в первую очередь неферментативное гликозилирование и оксидативный стресс, приводящие к модификациям генов и белков, задействованных в развитии диабетических микро- и макрососудистых осложнений [21–23]. Более того, достигнуто понимание того, какие именно эпигенетические механизмы ремоделирования хроматина, опосредованные гипергликемией, играют ключевую роль в изменении работы генов [24–26] (рис. 1).
/17/17.jpg)
Так, в настоящий момент постулируется, что избыток глюкозы, в том числе на уровне экспрессии соответствующих генов, меняет функционирование множества внутриклеточных путей, приводя к активации ряда изоформ протеинкиназы С, полиолового и гексозаминового пути, а также к усиленному образованию продуктов неферментативного гликозилирования, нарушению работы митохондрий (митохондриальной дисфункции) и программы апоптотической гибели клеток [22, 27]. Дисфункция митохондрий, в свою очередь, вызывает генерирование избытка активных форм кислорода и азота (оксидативный и нитрозивный стресс), приводя к ковалентным проатерогенным модификациям белков, липидов/липопротеинов и нуклеиновых кислот, формируя негативную клеточную память о гипергликемии [25, 28, 29]. С другой стороны, формируемая при СД резистентность тканей к инсулину, затрагивая главным образом метаболический сигналинг гормона и оставляя неизменным его ростовой сигналинг, добавляет проатерогенные стимулы на уровне генома, приводя к разрастанию гладкомышечных сосудистых клеток и хроническому воспалению [30].
В многочисленных экспериментальных работах последних лет показано, что оксидативный стресс, сопровождающий гипергликемию, вызывает активацию ряда так называемых стресс-реактивных транскрипционных факторов. Эти белковые молекулы могут быть разделены на 4 класса (с І по IV) в соответствии со структурными элементами, которые опосредуют ДНК-связывающую активность и определяют класс лекарственных средств, способных влиять на их функцию [31, 32]. Так, известно, что статины модулируют работу белка, связывающего стерол-реагирующий элемент — транскрипционный фактор І класса, регулирующий экспрессию генов, вовлеченных в метаболизм холестерина и жирных кислот.
Инсулин-сенситайзеры влияют на ядерный рецептор PPAR-гамма (активированный пролифератором пероксисом рецептор гамма) — транскрипционный фактор ІІ класса, контролирующий липидный и углеводный метаболизм, адипогенез и некоторые про-/антивоспалительные пути [32–35]. Фактически транскрипционные факторы семейства PPAR связывают липидные молекулы и регулируют транскрипцию генов-мишеней с привлечением коактиваторов и корепрессоров, как правило, гистоновых ацетилтрансфераз или гистоновых деацетилаз соответственно [33]. Их работа зависит от антиоксидантов, а природными лигандами являются омега-3 полиненасыщенные жирные кислоты и их метаболиты [36].
Однако наиболее изученным транскрипционным фактором с доказанным участием в процессах развития специфических и неспецифических сосудистых осложнений при СД 1-го и 2-го типа является ядерный фактор kВ (NF-kВ) — транскрипционный фактор ІV класса [25, 32, 37].
Так, на культуре эндотелиальных клеток аорты и на недиабетических мышах показано, что транзиторные (до 16 ч) пики гипергликемии вызывают долгосрочные активирующие эпигенетические изменения промотора субъединицы р65 в NF-kВ, что приводит к усилению экспрессии р65 и связанному с этим повышению синтеза хемоаттрактантного белка моноцитов 1 (МСР-1) и адгезивных молекул сосудистых клеток 1 (VCAM-1) в течение 6 суток после нормализации гликемии [21]. Характерно, что эти вызванные гипергликемией эпигенетические изменения предотвращались путем торможения митохондриальной сверхпродукции свободных радикалов или ингибированием образования конечных продуктов усиленного гликозилирования (таких как метилглиоксаль), подтверждая их участие в опосредовании передачи гипергликемического сигнала в геном. В то же время полученные результаты подчеркивают наличие длительных драматических проатерогенных последствий краткосрочной гипергликемии для сосудистых клеток и свидетельствуют о том, что гипергликемические подъемы могут быть независимым от уровня гликозилированного гемоглобина фактором риска развития диабетических осложнений.
Логично, что метаболический эпигенетический эффект не ограничивается только воздействием глюкозы. Так, в исследовании STENO 2 одновременный контроль уровней глюкозы, давления крови, липидного профиля и применение антитромботической терапии были связаны с 53% снижением риска макрососудистых осложнений у больных СД 2-го типа [38]. Результаты отдаленной фазы этого испытания подтвердили, что начальный жесткий метаболический контроль трансформируется в отсроченные положительные эффекты [39]. Исследование показало, что, несмотря на потерю после окончания активной фазы испытания различий в уровнях гликозилированного гемоглобина, кровяного давления и липидного профиля, через 5 лет сохранялись существенные различия по конечным микро- и макрососудистым точкам между группами с интенсивной и традиционной терапией.
Привлекает внимание тот факт, что положительный эффект метаболической памяти (называемый метаболическим наследием) более выражен, когда гликемический контроль достигается с самого начала развития заболевания. Это подтверждают данные, полученные при исследовании больных с СД как 1-го, так и 2-го типа, что позволило сформулировать концепцию о необходимости раннего интенсивного метаболического контроля, нацеленного на предотвращение развития диабетических осложнений. Исходя из этого предположения, было инициировано испытание ADDITION-Europe, целью которого стало сравнение интенсивной мультифакторной и общепринятой согласно национальным рекомендациям терапии пациентов с СД 2-го типа, впервые выявленного в результате скрининга [40]. По окончании 5-летнего проспективного исследования в обеих группах наблюдалось улучшение всех оцениваемых факторов сердечно-сосудистого риска, включая гликозилированный гемоглобин (6,6 против 6,7 %), диастолическое (80 против 81 мм рт.ст.) и систолическое (135 против 138 мм рт.ст.) давление, а также уровень холестерина липопротеинов низкой плотности (2,1 против 2,3 ммоль/л), со статистически недостоверным относительным снижением числа сердечно-сосудистых событий в группе с интенсивной терапией. Однако в данном исследовании авторы столкнулись с трудностями при формулировании конечных выводов. Это было связано с тем, что всем обследованным пациентам со времени постановки диагноза сразу было обеспечено качественное лечение, что привело к минимальным различиям выраженности факторов риска между группами и неожиданно низкому уровню сердечно-сосудистых событий, четко объясняемому оптимальным профилем факторов риска в обеих группах.
Более позднее сообщение Yang и соавт. [41] касается результатов исследования влияния интенсивной мультифакторной терапии пациентов с СД 2-го типа с длительностью заболевания менее 1 года и без клинических проявлений артериосклероза или субклинических атеросклеротических признаков. После 7-летнего периода лечения совокупная распространенность сердечно-сосудистых событий в группе интенсивной терапии была ниже, чем при общепринятой тактике лечения (10,00 и 32,35 % соответственно), подтверждая осуществимость первичной профилактики макрососудистых заболеваний у больных с небольшой длительностью СД 2-го типа.
/19/19.jpg)
К сожалению, эти исследования не предоставляют данных о длительности и выраженности метаболической памяти, сформированной при таком изначальном метаболическом контроле заболевания. Однако подобная информация будет собрана по окончании испытания ORIGIN [42]. В это исследование было рандомизировано 12 537 человек с факторами сердечно-сосудистого риска, нарушенной гликемией натощак, нарушенной толерантностью к глюкозе или СД 2-го типа для получения инсулина гларгин (с целевым уровнем гликемии натощак ≤ 5,3 ммоль/л) или стандартной терапии. Несмотря на тот факт, что это исследование не фокусировалось на оценке сравнения степени интенсивности снижения глюкозы, было обнаружено, что в группе инсулинотерапии средний уровень гликозилированного гемоглобина был ниже, чем при стандартной терапии (6,2 против 6,5 %) без существенного различия в показателях сердечно-сосудистых осложнений на момент окончания испытания (6,2 года). Все участники ORIGIN в настоящее время задействованы в продолжении исследования, названном ORIGINALE (ORIGIN and Legacy Effects) и направленном на оценку таких клинических событий, как нефатальный инфаркт миокарда и инсульт, смертность и другие ключевые показатели здоровья в течение последующих 8–9 лет [43].
Накопленные к настоящему времени доказательства эффективности раннего достижения уровней гликемии и гликозилированного гемоглобина, близких к нормальным, в предупреждении сердечно-сосудистых осложнений нашли свою реализацию в предложенном в январе 2014 года пересмотре рекомендаций Американской диабетической ассоциации относительно стандартов медицинской помощи при СД [44]. В частности, в разделе, касающемся фармакологической терапии гипергликемии при СД 2-го типа, сроки изменения алгоритма неинсулиновой терапии были сокращены с 3–6 до 3 мес. Так, больным с впервые диагностированным СД 2-го типа рекомендовано добавлять второй пероральный препарат, агонист глюкагоноподобного пептида 1 или инсулин в том случае, когда неинсулиновая монотерапия в максимально переносимой дозе не обеспечивает достижения или поддержания целевого уровня гликозилированного гемоглобина спустя 3 мес.
Кроме того, необходимо учитывать тот факт, что коррекция гипергликемии без уменьшения резистентности к инсулину может не оказать ни кардиопротекторного, ни антиатерогенного эффекта вследствие критических уровней инсулина, в особенности проинсулина, относящегося, по последним данным, к ведущим факторам сердечно-сосудистого риска [45, 46]. В связи с этим преимуществом обладают пероральные сахароснижающие средства с щадящей стимуляцией секреции инсулина (см. ниже) и инсулинотерапия.
Соотношение* «средний прирост инсулина в плазме крови (мкЕд/мл)/среднее (процентное) снижение глюкозы крови» составляет:
— глимепирид — 0,03;
— гликлазид — 0,07;
— глибенкламид — 0,16.
Издаваемые в последние годы рекомендации, основанные на новейших научных достижениях и всестороннем анализе крупных клинических и эпидемиологических исследований, все больше направлены на преодоление так называемой клинической инертности и требуют индивидуализированной терапевтической стратегии, учитывающей особенности больного и течение болезни, включая понимание патогенетической сложности заболевания и различные характеристики имеющейся на рынке фармакотерапии [44, 47].
Таким образом, обобщая весь имеющийся на настоящий момент фактический (экспериментальный и клинический) материал относительно вклада феномена гликемической памяти и эпигенетических модификаций в патогенез специфических и неспецифических сосудистых осложнений при СД 1-го и 2-го типа, можно сделать простой и всеобъемлющий вывод: для диабетических пациентов максимально безопасное достижение уровней гликемии и гликозилированного гемоглобина, близких к нормальным, с момента постановки диагноза, а в идеале — и его поддержание в течение максимально возможного срока является критическим для успешной профилактики микро- и макрососудистых осложнений. Это не только поможет предупредить/замедлить развитие диабетических осложнений, но и создаст позитивное «метаболическое наследие», а также предотвратит накопление негативной метаболической/гликемической памяти.
1. Diabetes mellitus, fasting glucose, and risk of cause-specific death / S.R. Seshasai, S. Kaptoge, A. Thompson [et al.]; The Emerging Risk Factors Collaboration // N. Engl. J. Med. — 2011. — Vol. 364. — P. 829–841.
2. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus / D.M. Nathan, J. Lachin, P. Cleary [et al.]; Diabetes Control and Complications Trial; Epidemiology of Diabetes Interventions and Complications Research Group // N. Engl. J. Med. — 2003. — Vol. 348. — P. 2294–2303.
3. Diabetes control and complications trial/epidemiology of diabetes interventions and complications research group. Prolonged effect of intensive therapy on the risk of retinopathy complications in patients with type 1 diabetes mellitus. Ten years after the diabetes control and complications trial [Text] // Arch. Ophtalmol. — 2008. — Vol. 126. — P. 1707–1715.
4. Vibration perception threshold as a measure of distal symmetrical peripheral neuropathy in type 1 diabetes: results from the DCCT/EDIC study [Text] / C.L. Martin, B.H. Waberski, R. Pop-Busui [et al.] // Diabetes Care. — 2010. — Vol. 33. — P. 2635–2641.
5. Association between cardiovascular autonomic neuropathy and left ventricular dysfunction in the diabetes control and complications trial/epidemiology of diabetes interventions and complications (DCCT/EDIC) study [Text] / R. Pop-Busui, P.A. Clearly, B.H. Braffett [et al.] // J. Am. Coll. Cardiol. — 2013. — Vol. 61. — P. 447–454.
6. Intensive diabetes therapy and glomerular filtration rate in type 1 diabetes [Text] / I.H. De Boer, W. Sun, P.A. Clear [et al.] // N. Engl. J. Med. — 2011. — Vol. 365. — P. 2366–2376.
7. UKPDS Group. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study // BMJ. — 2000. — Vol. 321. — P. 405–412.
8. 10-year follow-up of intensive glucose control in type 2 diabetes / R.R. Holman, S.K. Paul, M.A. Bethel [et al.] // N. Engl. J. Med. — 2008. — Vol. 359. — P. 1577–1589.
9. The ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with Type 2 Diabetes // N. Engl. J. Med. — 2008. — Vol. 358. — P. 2560–2572.
10. Intensive blood glucose control and complications in American veterans with type 2 diabetes [Text] / W. Duckworth, C. Abraria, T. Moritz [et al.] // N. Engl. J. Med. — 2009. — Vol. 360. — P. 129–139.
11. Effects of intensive glucose lowering in type 2 diabetes [Text] / H.C. Gerstein, M.E. Miller, R.P. Byington [et al.] // N. Engl. J. Med. — 2008. — Vol. 358. — P. 2545–2559.
12. Engerman R.L. Progression of incipient diabetic retinopathy during good glycemic control / R.L. Engerman, T.S. Kern // Diabetes. — 1987. — Vol. 36. — P. 808–812.
13. Kowluru R.A. Effect of re-institution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats // Diabetes. — 2003. — Vol. 52. — P. 818–823.
14. Kowluru R.A. Reversal of hyperglycemia and diabetic nephropathy: effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats / R.A. Kowluru, S.N. Abbas, S. Odenbach // J. Diabetes Complicat. — 2004. — Vol. 18. — P. 282–288.
15. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail / D. Brasacchio, J. Okabe, M.E. Cooper, A. El–Osta // Diabetes. — 2009. — Vol. 58. — P. 1229–1236.
16. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory / S. Roy, R. Sala, E. Cagliero, M. Lorenzi // Proc. Natl. Acad. Sci. USA. — 1990. — Vol. 87. — P. 404–408.
17. Holliday R. Epigenetics. A historical overview [Text] // Epigenetics. — 2006. — Vol. 1, № 2. — Р. 76–80.
18. MicroRNA-29b induces global DNA hypomethylation and tumor suppressоr gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1 [Text] / R. Garzon, S. Liu, M. Fabbri [et al.] // Blood. — 2009. — Vol. 113. — P. 6411–6418.
19. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development [Text] // Nature. — 2007. — Vol. 447. — P. 425–432.
20. Epigenetic differences arise during the lifetime of monozygotic twins [Text] / M.F. Fraga, E. Ballestar, M.F. Paz [et al.] // PNAS. — 2005. — Vol. 102. — P. 10604–10609. doi: 10.1073/pnas.0500398102.
21. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia [Text] / A. El–Osta, D. Brasacchio, D. Yao [et al.] // J. Exp. Med. — 2008. — Vol. 205. — P. 2409–2417.
22. Brownlee M. The pathobiology of diabetes complications: a unifying mechanism [Text] // Diabetes. — 2005. — Vol. 54. — P. 1615–1625.
23. Stitt A.W. The role of advanced glycation in the pathogenesis of diabetic retinopathy [Text] // Exp. Mol. Pathol. — 2003. — Vol. 1. — P. 95–108.
24. Siebel A.L. Glycemic memory associated epigenetic changes [Text] / A.L. Siebel, A.Z. Fernandez, A. El-Osta // Biochem. Pharmacol. — 2010. — Vol. 80. — P. 1853–1859.
25. Cooper M. Epigenetics: mechanisms and implications for diabetic complications [Text] / M. Cooper, A. El-Osta // Circ. Res. — 2010. — Vol. 107. — P. 1407–1413.
26. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: bringing all the strands together [Text] / F. Paneni, M. Volpe, T.F. Luscher, F. Cosentino // Diabetes. — 2013. — Vol. 62. — P. 1800–1807. doi: 10.2337/db12-1648.
27. Brownlee M. Biochemistry and molecular cell biology of diabetic complications [Text] // Nature. — 2001. — Vol. 414. — P. 813–820.
28. El Osta A. Glycemic memory [Text] // Curr. Opin. Lipidol. — 2012. — Vol. 23. — P. 24–29. doi: 10.1097/MOL.0b013e32834f319d
29. Reactive oxygen species mediate a cellular «memory» of high glucose stress signaling [Text] / M.A. Ihnat, J.E. Thorpe, C.D. Camat [et al.] // Diabetologia. — 2007. — Vol. 50. — P. 1523–1531.
30. DeFronzo R.A. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus [Text] // Diabetes. — 2009. — Vol. 58. — P. 773–795.
31. Bernstein B.E. The mammalian epigenome [Text] / B.E. Berstein, A. Meissner, E.S. Lander // Cell. — 2007. — Vol. 128. — P. 669–681.
32. De Nigris F. New insights in the transcriptional activity and coregulator molecules in the arterial wall [Text] / F. de Nigris, L.O. Lerman, C. Napoli // Int. J. Cardiol. — 2002. — Vol. 86. — P. 153–168.
33. Plutzki J. The PPAR–RXR transcriptional complex in the vasculature: energy in the balance [Text] // Circ. Res. — 2011. — Vol. 108. — P. 1002–1016. doi: 10.1161/CIRCRESAHA.110.226860.
34. Role of peroxisome proliferation-activated receptor-γ in atherosclerosis: an update [Text] / N. Wang, R. Yin, Y. Liu [et al.] // Circ. J. — 2011. — Vol. 75. — P. 528–535. doi: 10.1253/circj.CJ-11-0060.
35. Lund G. Atherosclerosis: An epigenetic balancing act that goes wrong [Text] / G. Lund, S. Zaina // Curr. Atheroscler. Rep. — 2011. — Vol. 13. — P. 208–214. doi: 10.1007/s11883-011-0174-3.
36. De Santa Olalla L.M. N-3 fatty acids in glucose metabolism and insulin sensitivity [Text] / L.M. de Santa Olalla, F.J.S. Muniz, M.P. Vaquero // Nutr. Hosp. — 2009. — Vol. 24. — P. 113–127.
37. NF-kappaB regulation: the nuclear response [Text] / A.K. Mankan, M.W. Lawless, S.G. Gray [et al.] // J. Cell. Mol. Med. — 2009. — Vol. 13. — P. 631–643. doi: 10.1111/j.1582–4934.2009.00632.x.
38. Multifactorial intervention and cardiovascular diseases in patients with type 2 diabetes [Text] / P. Gaede, P. Vedel, N. Larsen [et al.] // N. Engl. J. Med. — 2003. — Vol. 348. — P. 383–393. doi: 10.1056/NEJMoa021778.
39. Effect of a multifactorial intervention oh mortality in type 2 diabetes [Text] / P. Gaede, H. Lund-Andersen, H.H. Parving [et al.] // N. Engl. J. Med. — 2008. — Vol. 358. — P. 580–591. doi: 10.1056/NEJMoa0706245.
40. Effect of early intensive multifactorial therapy on 5-year cardiovascular outcomes in individuals with type 2 diabetes detected by screening (ADDITION Europe): a cluster-randomized trial [Text] / S.J. Griffin, K. Borch-Johnsen, M.J. Davies [et al.] // Lancet. — 2011. — Vol. 378. — P. 156–167. doi: 10.1016/S0140–6736(11)60698–3.
41. Primary prevention of macroangiopathy in patients with short-duration type 2 diabetes by intensified multyfactorial intervention: seven-year follow-up of diabetes complications in Chinese [Text] / Y. Yang, I.J. Yao, L.J. Du [et al.] // Diabetes Care. — 2012. doi: 10.2337/dc12-0227.
42. Basal insulin and cardiovascular and other outcomes in dysglycemia [Text] / H.C. Gerstein, J. Bosch, G.R. Dagenais [et al.] // N. Engl. J. Med. — 2012. — Vol. 367. — P. 319–328. doi: 10.1056/NEJMoa1203858.
43. The ORIGINALE Study. http://origintrial.org/Assets/PDF/ORIGINALE%20Protocol.pdf
44. Summary of revisions to the 2014 clinical practice recommendations [Text] / American Diabetes Association // Diabetes Care. — 2014. — Vol. 37, Suppl. 1. — P. S4–S80. doi: 10.2337/dc14-S004.
45. Circulating concentrations of insulin markers and coronary heart disease: a quantitative review of 19 Western prospective studies [Text] / N. Sarwar, N. Sattar, V. Gudnason, J. Danesh // Eur. Heart J. — 2007. — Vol. 28. — P. 2491–2497. doi: 10.1093/eurheartj/ehm115.
46. Jandeleit-Dahm K.A. Insulin and cardiovascular disease: biomarker or association? [Text] / K.A. Jandeleit-Dahm, S.P. Gray // Diabetologia. — 2012. — Vol. 55. — P. 3145–3151. doi: 10.1007/s00125–012–2729–4.
47. Management of hyperglycemia in type 2 diabetes: a patient-centered approach. Position Statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) [Text] / S.E. Inzucchi, R.M. Bergenstal, J.B. Buse [et al.] // Diabetes Care. — 2012. — Vol. 35. — P. 1–16. doi: 10.2337/dc12-0413.