
Международный эндокринологический журнал 1 (65) 2015
Вернуться к номеру
Патоморфологічні зміни підшлункової залози та роль судинного фактора у розвитку інсулінової недостатності при цукровому діабеті 2-го типу
Авторы: Вернигородський С.В. — Вінницький національний медичний університет імені М.І. Пирогова
Рубрики: Эндокринология
Разделы: Клинические исследования
Версия для печати
В статті наведені сучасні дані щодо патогенетичних ланок виникнення цукрового діабету (ЦД) 2-го типу. Проведено морфологічне дослідження судин підшлункової залози у хворих на ЦД 2-го типу. Виявлені специфічні зміни в них у вигляді генералізованого гіалінозу, що характерно для діабетичних ангіопатій. За нашими даними, саме ураження судин відіграє ключову роль у порушенні кровопостачання бета-клітин, що призводить до дегенеративних процесів, прогресування інсулінової недостатності та погіршення перебігу ЦД 2-го типу.
В статье представлены современные данные о патогенетических звеньях возникновения сахарного диабета (СД) 2-го типа. Проведено морфологическое исследование сосудов поджелудочной железы у больных СД 2-го типа. Обнаружены специфические изменения в них в виде генерализованного гиалиноза, что характерно для диабетических ангиопатий. По нашим данным, именно поражение сосудов играет ключевую роль в нарушении кровоснабжения бета-клеток, что приводит к дегенеративным процессам, прогрессированию инсулиновой недостаточности и ухудшению течения СД 2-го типа.
The article presents current data on pathogenetic links of diabetes mellitus (DM) type 2 occurrence. A morphological study of pancreas vessels in patients with DM type 2 has been carried out. Specific changes in them as generalized hyalinosis, which is characteristic of diabetic angiopathy, were identified. Our records show that exactly vascular lesions has a key role in circulatory disorders of beta-cells, leading to degenerative processes, progression of insulin deficiency and worsening of DM type 2 course.
цукровий діабет, судини підшлункової залози, морфологічні зміни.
cахарный диабет, сосуды поджелудочной железы, морфологические изменения.
diabetes mellitus, pancreas vessels, morphological changes.
Статья опубликована на с. 27-30
Цукровий діабет (ЦД) — одна з глобальних та актуальних проблем сучасної медицини. Це пов’язано з неухильним ростом його частоти, з одного боку, та тими тяжкими серцево-судинними ускладненнями, що призводять до ранньої інвалідизації та смертності — з іншого. На сьогодні відомо, що ЦД 2-го типу — гетерогенне захворювання, основу якого становить інсулінорезистентність (ІР) і недостатність функції бета-клітин. Встановлено, що спочатку у генетично схильних до ЦД 2-го типу осіб виникає і прогресує ІР. ІР м’язової тканини розглядається як найбільш ранній і, можливо, генетично обумовлений дефект, який набагато випереджує появу гіперглікемії [1]. Однак відсутня єдина думка стосовно того, яке з цих порушень є первинним у розвитку захворювання. Усі існуючі концепції етіології та патогенезу ЦД виходять з поняття первинного порушення діяльності інсулярного апарату внаслідок поєднання генетичних, імунологічних, інфекційних та інших патологічних чинників, що призводять до зниження секреції (або біологічної дії) інсуліну та розвитку всього симптомокомплексу захворювання, провідним з яких є підвищення вмісту цукру в крові. Останнє за умов абсолютної або відносної недостатності інсуліну обумовлене, з одного боку, зниженням утилізації глюкози тканинами, а з іншого — збільшенням її продукції в кров за рахунок активації механізмів глікогенолізу і глюконеогенезу. Виникнення та розвиток судинних і органних ускладнень при ЦД у першу чергу асоціюється з наявністю хронічної гіперглікемії. Виходячи з цього, традиційно проведене лікування спрямоване головним чином на компенсацію недостатності ендогенного інсуліну з відновленням нормоглікемії, що вважається заходом профілактики різноманітних ускладнень ЦД [2].
Разом з тим механізм, відповідальний за прогресуюче зниження функції бета-клітин, до кінця не з’ясований. Є декілька гіпотез, які показують, що збільшення частоти апоптозу та зниження регенерації бета-клітин генетично запрограмовані. Крім того, надлишкова секреція інсуліну в ранній період інсулінорезистентності може призводити до збільшення загибелі клітин, а накопичення аміліну — до амілоїдозу острівців [3–8]. Порушення ліпідного обміну і надлишкове накопичення неестерифікованих жирних кислот (НЕЖК) в острівцях підшлункової залози також може призводити до зниження їх функціональної активності, так званої ліпотоксичності [4]. Значним проривом у розумінні механізмів деструкції бета-клітин підшлункової залози cтали дослідження біологічної значущості оксиду азоту (NO). Дослідженнями останніх років показано, що саме оксид азоту, який утворюється в острівцях і бета-клітинах підшлункової залози, відіграє основну роль у механізмах руйнування і загибелі бета-клітин, що і призводить до різкого зменшення їх кількості та розвитку клінічного ЦД [9–11]. Не викликає сумніву той факт, що ангіогенні фактори, такі як судинний ендотеліальний фактор росту, фактор росту фібробластів та інші, відіграють ключову роль у розвитку мікросудинних ускладнень при ЦД [12]. Разом із тим у літературі практично відсутні роботи, які були б присвячені вивченню судинного фактора у розвитку інсулінової недостатності при ЦД 2-го типу. Тому метою нашого дослідження було вивчення патоморфологічних змін судин підшлункової залози при даній патології.
Матеріали та методи дослідження
Патоморфологічні дослідження підшлункової залози виконані 46 померлим на ЦД 2-го типу віком від 20 до 75 років (табл. 1) та тривалістю захворювання від 1 до 20 років.
/28/28.jpg)
Тривалість захворювання на ЦД 2-го типу у досліджуваних осіб становила: до 5 років — 8 осіб, від 6 до 10 років — 14, від 10 до 20 років — 16, понад 20 років — 8 осіб. Смерть хворих настала від інфаркту міокарда (18), ішемічного інсульту (10), тромбоемболії легеневої артерії (4), серцево-судинної недостатності після операції ампутації нижніх кінцівок із приводу гангрени стопи (6) і ниркової недостатності (8). Контрольну групу становли 14 померлих аналогічного віку без порушення вуглеводного обміну. Шматочки тканин фіксували у 5% розчині нейтрального формаліну протягом 24–48 годин, зневоднювали в спиртах зростаючої міцності та заливали в парафін. З парафінових блоків були зроблені серійні зрізи завтовшки 5–8 мкм, що забарвлювали гематоксилін-еозином, за Ван Гізоном, толуїдиновим синім. Окремі гістологічні препарати фарбували суданом III, IV, орсеїном та імпрегнували азотнокислим сріблом за Футом та Гоморі, резорцин-фуксином за Вейгертом. З метою диференціювання альфа- та бета-клітин підшлункової залози застосовували гематоксилін з фуксином за Гоморі.
Результати та їх обговорення
Макроскопічні зміни підшлункової залози у хворих на ЦД 2-го типу відзначалися в усіх вікових групах та були більш виражені в осіб з тривалістю ЦД 10–20 та більше років. Макроскопічно спостерігали ліпоматоз підшлункової залози, який переважав у 54 % померлих, атрофію — у 41 % та склероз — у 40 %. У більшості випадків підшлункова залоза була у вигляді тонкого щільного тяжу розмірами від 11 до 16,5 см, товщиною 2–2,5 см. Тканина на розрізі малокрівна, щільноеластичної консистенції. Рисунок часточок був нечітким унаслідок дифузного розростання волокнистої світло-сірої сполучної тканини у вигляді сітки та вогнищевого розростання жирової тканини, яка інколи повністю заміщувала хвостовий відділ залози. Фіброзна капсула була склерозована. Місцями сполучна тканина у вигляді пучків сірого кольору розросталася навколо протоків і судин. Просвіти панкреатодуоденальних артерій на 1/3–1/4 перекриті ліпідними бляшками як в проксимальних, так і в дистальних відділах.
При патогістологічному дослідженні строма залози у вигляді тонких прошарків сполучної тканини з помірно повнокровними кровоносними судинами. Екскреторний апарат був представлений різної величини та форми залозистими часточками, між якими траплялися вивідні протоки різного калібру, вистелені однорядним призматичним і кубічним епітелієм. Інкреторний відділ представлений острівцями Лангерганса, що розташовані між ацинусами. Острівцеві альфа-клітини були нечисленні, локалізувалися переважно в периферичних відділах острівців, неаргірофільні, при забарвленні за Гоморі містять у цитоплазмі зернистість червоного кольору. Бета-клітини овальної форми, добре імпрегнуються солями срібла, цитоплазма їх дрібнозерниста, синюватого відтінку, розташовані вони переважно в центральних відділах острівців.
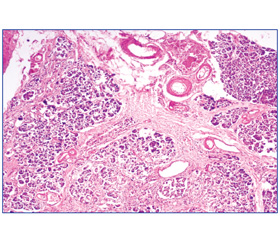
При гістологічному дослідженні на тлі нормальних часточок з незміненими ацинусами, клітини яких багаті на зимоген, чітко визначаються часточки, де клітини ацинусів дискомплексовані, з незначною кількістю зимогену, гранули якого розташовуються в базальних відділах клітин, цитоплазма їх вакуолізована, багата ліпідами. Строма органа представлена розростанням широких пластів сполучної тканини, різного ступеня зрілості, переважно навколо вивідних протоків, яка проростає між часточками та вростає в їх центральні відділи, що призводить до атрофії та формування несправжніх часточок (рис. 1).
Епітелій протоків сплощений, місцями десквамований. Поряд із дифузним склерозом спостерігали надлишкове розростання жирової тканини, яка заповнювала як прошарки між часточками, так і вростала всередину часточок. Кількість острівців Лангерганса значно зменшена, вони овальної або неправильної форми, оточені ніжною сполучнотканинною капсулою, що зливається з проміжною сполучною тканиною. В окремих випадках вони були представлені у вигляді груп дрібних клітин, погано обмежених від сусідньої тканини. Окремі острівці компенсаторно гіпертрофовані. Співвідношення між альфа- та бета-клітинами порушено в бік альфа-клітин. У бета-клітинах знаходили дегрануляцію цитоплазми, а у випадках з тяжким перебігом ЦД — гідропічну дегенерацію. Такі клітини мали світлу («пусту») цитоплазму, в якій гранули заміщені вакуолями. У 39 % випадків відзначали гіаліноз острівців із субендотеліальним відкладанням гомогенного ацидофільного гіаліну. Одночасно з гіалінозом спостерігалася і жирова інфільтрація бета-клітин. З боку судинного русла відзначається виражена перебудова як артеріальних, так і венозних судин у вигляді перекалібрування їх просвіту за рахунок плазматичного просякнення стінки, гіалінозу, склерозу внутрішньої оболонки. Так, по ходу судин дрібного калібру спостерігалися однорідні склоподібні тяжі, гіаліноз їх стінок. Слід відзначити, що процес розпочинався із скупчення у просвіті судин еозинофільної білкової маси з подальшою плазморагією стінки (рис. 2).
/29/29.jpg)
У міжчасточкових артеріолах поряд із гіалінозом стінок спостерігали виражений склероз із звуженням просвіту судин понад 75 %.
У панкреатодуоденальних артеріях відзначали наявність ліпідно-фіброзних та гіалінізованих бляшок, подібних до тих, що знаходили в коронарних артеріях у хворих з ішемічною хворобою серця та артеріальною гіпертензією, які звужували просвіт судин на 1/3–1/4.
Отже, результати проведеного морфологічного дослідження показали, що при ЦД 2-го типу поряд з ліпоматозом, склерозом та атрофією підшлункової залози, які знаходили в усіх вікових групах, у стінках кровоносних судин розвиваються зміни як клітинних, так і волокнистих структур.
Привертає увагу плазморагія та накопичення гіаліну у стінках судин мікроциркуляторного русла. Такий генералізований гіаліноз, проявом якого стає макро- і мікроангіопатія, слід розглядати як специфічні діабетичні прояви у тканині підшлункової залози. Майже в усіх випадках у підшлунковій залозі виявляються явища гіалінозу, що вкрай рідко трапляється при ЦД 1-го типу. Як відомо, ознакою функціональної недостатності бета-клітин при ЦД 2-го типу є зменшення розміру ядер клітин, комплексу Гольджі та ендоплазматичної сітки, що, найімовірніше, пов’язане з розвитком склерозу судин, фіброзу і гіалінозу острівців [10]. Ці зміни виявляються більшою чи меншою мірою в підшлунковій залозі осіб похилого віку за відсутності порушення вуглеводного обміну. Недостатність бета-клітин при ЦД 2-го типу може бути наслідком зниження чутливості цих клітин до глюкози, порушення процесів входження кальцію в бета-клітину, його зв’язування з кальмодуліном, зміни мікротубулярної-мікроворсинчастої системи, дефекту фібрилярних білків цитоплазми.
Патоморфологічні зміни судин підшлункової залози, на нашу думку, призводять до порушення кровопостачання бета-клітин, розвитку в них дистрофічних і дегенеративних процесів і, як наслідок, зменшення секреції інсуліну з подальшим прогресуванням інсулінової недостатності та погіршенням перебігу ЦД 2-го типу.
Отже, дані огляду літератури та результати нашого дослідження підтверджують безсумнівну участь ангіогенних факторів у механізмах формування та прогресування ЦД 2-го типу.
Висновки
1. Морфологічні зміни в судинах підшлункової залози носять специфічний характер для ЦД 2-го типу і характеризуються генералізованим гіалінозом.
2. Структурно змінені судини підшлункової залози призводять до порушення кровопостачання бета-клітин, розвитку в них дистрофічних і дегенеративних процесів, що сприяє прогресуванню інсулінової недостатності.
3. Подальше патоморфологічне дослідження ультраструктурних змін у судинному руслі підшлункової залози сприятиме вивченню більш тонких механізмів розвитку ЦД 2-го типу та відокремленню диференційно-діагностичних критеріїв між діабетичними макро- та мікроангіопатіями і ураженнями судин при ішемічній хворобі серця та артеріальній гіпертензії.
1. Полторак В.В. Гликемическая память как патогенетическое основание для формирования алгоритма современной антидиабетической терапии / В.В. Полторак, Н.С. Красова, М.Ю. Горшунская // Международный эндокринологический журнал. — 2014. — № 3(59). — С. 15–21.
2. Полторак В.В. Адипонектин та цукровий діабет 2-го типу (патогенетичні аспекти як підґрунтя для оптимізації антидіабетичної фармакотерапії) [Текст] / В.В. Полторак, М.Ю. Горшунська, Н.С. Красова // Международный эндокринологический журнал. — 2014. — № 5(61). — С. 95–104.
3. Lidell M.E., Betz M.J., Enerbаck S. Brown adipose tissue and its therapeutic potential // J. Intern. Med. — 2014. — Vol. 276(4). — P. 364–377. — Available from: http://doi.wiley.com/10.1111/joim.12255 doi: 10.1111/joim.12255.
4. Samuel V.T., Petersen K.F., Shulman G.I. Lipid-induced insulin resistance: unravelling the mechanism // Lancet. — 2010. — Vol. 375(9733). — P. 2267–2277. — Available from: http://linkinghub.elsevier.com/retrieve/pii/S0140673610604084 doi: 10.1016/S0140-6736(10)60408-4.
5. Grayson B.E., Seeley R.J., Sandoval D.A. Wired on sugar: the role of the CNS in the regulation of glucose homeostasis // Nat. Rev. Neurosci. — 2013. — Vol. 14(1). — P. 24–37. — Available from: http://www.nature.com/doifinder/10.1038/nrn3409 PubMed PMID: 23232606. doi: 10.1038/nrn3409.
6. Hotamisligil G.S. Inflammation and metabolic disorders // Nature. — 2006. — Vol. 444(7121). — P. 860–867. — Available from: http://www.nature.com/doifinder/10.1038/nature05485 PubMed PMID: 17167474. doi: 10.1038/nature05485.
7. Kadowaki T., Yamauchi T. Adiponectin Receptor Signaling: A New Layer to the Current Model. Cell // Metabolism. — 2011. — Vol. 13(2). — P. 123–124. — Available from: http://linkinghub.elsevier.com/retrieve/pii/S1550413111000131 PubMed PMID: 21284979. doi: 10.1016/j.cmet.2011.01.012.
8. Goldfine A.B., Kahn C.R. Adiponectin: linking the fat cell to insulin sensitivity // Lancet. — 2003. — Vol. 362(9394). — P. 1431–1432. — Available from: http://linkinghub.elsevier.com/retrieve/pii/S0140673603147277. doi: 10.1016/S0140-6736(03)14727-7.
9. Holland W.L., Miller R.A., Wang Z.V. et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin // Nat. Med. — 2010. — Vol. 17(1). — P. 55–63. Available from: http:// www.nature.com/doifinder/10.1038/nm.2277 PubMed PMID: 21186369. doi: 10.1038/nm.2277.
10. Fabbrini E., Magkos F., Mohammed B.S. et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity // Proceedings of the National Academy of Sciences. — 2009. — Vol. 106(36). — P. 15430–15435. — Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0904944106 PubMed PMID: 19706383. doi: 10.1073/pnas.0904944106.
11. Morino K., Petersen K.F., Shulman G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction // Diabetes. — 2006. — Vol. 55(Suppl. 2). — P. 9–15. — Doi: 10.2337/db06-S002.
12. Penkov D.N., Egorov A.D., Mozgovaya M.N., Tkachuk V.A. Insulin resistance and adipogenesis: Role of transcription and secreted factors // Biochemistry. — 2013. — Vol. 78(1). — P. 8–18. — Available from: http:// link.springer.com/ 10.1134/S0006297913010021 PubMed PMID: 23379555. doi: 10.1134/S0006297913010021.