
Международный эндокринологический журнал 1 (65) 2015
Вернуться к номеру
Лабораторна діагностика окремих компонентів метаболічного синдрому
Авторы: Кобиляк Н.М. — Національний медичний університет імені О.О. Богомольця, м. Київ; Кирієнко Д.В. —
Київський міський клінічний ендокринологічний центр
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
Метаболічний синдром являє собою комплекс взаємопов’язаних факторів ризику розвитку серцево-судинних захворювань та цукрового діабету. Поширеність метаболічного синдрому прогресивно зростає у всьому світі внаслідок збільшення чисельності людей з ожирінням і неправильним способом життя. У статті наведено визначення поняття інсулінорезистентності та порушення ендокринної функції жирової тканини як основних патогенетичних ланок, що об’єднують основні кластери метаболічного синдрому. Продемонстровано, що метаболічний синдром є комплексною медичною проблемою, рішення якої повинне бути спрямоване на зниження поширеності ожиріння, підвищення фізичної активності населення та раннє виявлення з метою модифікації факторів ризику, які призводять до розвитку цукрового діабету типу 2 та серцево-судинних захворювань.
Метаболический синдром представляет собой комплекс взаимосвязанных факторов риска развития сердечно-сосудистых заболеваний и сахарного диабета. Распространенность метаболического синдрома прогрессивно растет по всему миру вследствие увеличения численности людей с ожирением и неправильным образом жизни. В статье представлено определение понятия инсулинорезистентности и нарушения эндокринной функции жировой ткани как основных патогенетических звеньев, которые объединяют основные кластеры метаболического синдрома. Продемонстрировано, что метаболический синдром является комплексной медицинской проблемой, решение которой должно быть направлено на снижение распространенности ожирения, повышение физической активности населения и раннее выявление с целью модификации факторов риска, которые приводят к развитию сахарного диабета типа 2 и сердечно-сосудистых заболеваний.
Metabolic syndrome is a complex of interrelated risk factors for cardiovascular diseases and diabetes mellitus. The prevalence of metabolic syndrome grows progressively all over the world due to the increased number of people with obesity and unhealthy lifestyle. The article presents the definition of insulin resistance and disorders in endocrine function of adipose tissue as major pathogenetic links that unite the main clusters of metabolic syndrome. It is shown that metabolic syndrome is a complex medical problem which should to be aimed at reducing the prevalence of obesity, increased physical activity of population and early detection in order to modify risk factors leading to the development of type 2 diabetes mellitus and cardiovascular diseases.
метаболічний синдром, інсулінорезистентність.
метаболический синдром, инсулинорезистентность.
metabolic syndromes, insulin resistance.
Статья опубликована на с. 73-84
Метаболічний синдром (МС) являє собою комплекс взаємопов’язаних факторів ризику розвитку серцево-судинних захворювань (ССЗ) та цукрового діабету (ЦД). Вперше у 1988 р. термін «метаболічний синдром Х» запропонував Reaven, у якому поєднав інсулінорезистентність (ІР), гіперінсулінемію, порушення толерантності до глюкози (ПТГ), дисліпопротеїнемію, гіпертригліцеридемію та артеріальну гіпертензію (АГ) [1]. Kaplan у 1989 р. як «смертельний квартет» охарактеризував поєднання абдомінального ожиріння, порушеної толерантності до глюкози, АГ та гіпертригліцеридемії [2]. З середини 90-х рр. починає переважати термін «метаболічний синдром», запропонований Henefeld у 1980 р., ще до опублікування Reaven його концепції [3].
Уже кілька десятиліть увага вчених зосереджена на ключовій ролі ІР як сполучної ланки серед патологічних факторів, що об’єднуються поняттям МС, проте її патогенез та чіткі діагностичні критерії даного синдрому залишаються до кінця нез’ясованими [4]. Продемонстровано, що поширеність МС прогресивно зростає у всьому світі внаслідок збільшення чисельності людей з ожирінням і неправильним способом життя. Ожиріння є незалежним фактором ризику розвитку ССЗ і смертності в загальній популяції [5, 6]. Деякі автори серед людей, які страждають від ожиріння, виділяють категорію пацієнтів із низьким ризиком розвитку серцево-судинних подій — так зване метаболічно здорове ожиріння (МЗО, metabolically normal or healthy obesity) [7–9]. МЗО характеризується відсутністю будь-якого явного кардіометаболічного захворювання — ЦД 2-го типу (ЦД2), дисліпідемії й АГ в осіб з індексом маси тіла (ІМТ) > 30 кг/м2. Додатковим критерієм, окрім кластерів МС, що використовується для підтвердження МЗО, є незначна вираженість синдрому хронічної системної запальної відповіді. За даними різних авторів, поширеність МЗО в популяції хворих на ожиріння становить 20–30 % [10]. У попередніх короткотривалих дослідженнях продемонстровано, що даний фенотип ожиріння асоційований із зменшенням ризику розвитку ЦД2 і CCЗ [11].
Поняття «предіабет» включає такі стани, як порушена глікемія натще та ПТГ. Даний термін був запропонований департаментом здоров’я США у 2002 році з метою привернення уваги громадськості до цієї проблеми, яка набула соціального значення та підкреслює високий ризик розвитку ЦД2 в подальшому (приблизно 10 % випадків на рік) [12]. Дані ВООЗ і ADA (Американська діабетична асоціація) свідчать про те, що у 27 % осіб із нормальними показниками глюкози натще може розвинутись предіабет і у 8 % — ЦД, а за наявності попереднього порушення глікемії в 50 % осіб виникають клінічні ознаки ЦД, причому тривалість періоду трансформації перебуває в межах від 3 до 10 років [13].
Для діагностики предіабету використовуються вимірювання рівня глікемії натще і пероральний глюкозотолерантний тест. У минулому тест на визначення глікованого гемоглобіну (HbA1c) використовувався лише з метою контролю рівня глюкози в крові, але не для встановлення діагнозу. Проте після стандартизації методу визначення, починаючи з 2009 року, міжнародна комісія експертів рекомендувала його використання для діагностики ЦД2 та предіабету [14]. ADA у своїх рекомендаціях (2010) для діагностики предіабету ввела додатковий індикатор — пограничний рівень HbA1c (5,7–6,4 %), який є інтегральним показником плазмового рівня глюкози [15]. Згідно з даними рекомендацій, пацієнтам із діагнозом «предіабет» рекомендовано проведення повторного визначення HbA1c протягом одного року. Діагноз предіабету також не варто виключати в людей із HbA1c < 5,7 % за наявності інших факторів ризику, а пацієнтів із рівнем HbA1c > 6,0 % слід зарахувати до групи високого ризику розвитку ЦД2.
Таким чином, МС є комплексною медичною проблемою, вирішення якої повинне включати зниження поширеності ожиріння, підвищення фізичної активності населення та раннє виявлення з метою модифікації факторів ризику, що призводять до розвитку ЦД2 та ССЗ.
Існуючі уявлення про патогенез МС укладаються в рамки трьох теорій. Першою з них була глюкоцентрична. Наприкінці 80-х рр. минулого століття їй на зміну прийшла ліпоцентрична теорія. Нарешті, у даний час найбільш бурхливо розвиваються дослідження в руслі ліпокінової теорії МС.
Згідно з глюкоцентричною гіпотезою, в основі розвитку МС лежить єдиний патологічний процес — інсулінорезистентність периферичних тканин, наслідком якого є гіперінсулінемія. Саме з ІР і супутньою їй гіперінсулінемією пов’язують усі метаболічні розлади, що спостерігаються при МС.
Згідно з двома іншими гіпотезами, ключову роль у розвитку МС відіграє вісцеральна жирова тканина. Абдомінальне (андроїдне) ожиріння розглядається як «генератор» і один з основних кластерів МС. Проте сформувались дві школи з принципово різними поглядами на роль вісцерального ожиріння в патогенезі МС [16].
Представники першої і більш давньої теорії дотримуються «портальної гіпотези» (ліпоцентрична теорія), яка була висунута Bjоrntorp [17]. В її основі лежить надлишок вільних жирних кислот (ВЖК), які утворюються в результаті вивільнення їх з адипоцитів за участю гормоночутливої ліпази та ліпопротеїнової ліпази (ЛПЛ) на ендотелії капілярів легень, серця та інших внутрішніх органів. Швидкість ліполізу як in vitro, так і in vivo залежить як від активності ліпази адипоцитів, так і від функціональної рівноваги між ліполітичними та антиліполітичними регуляторами. Пропорційно збільшенню розміру адипоцита зростає активність ЛПЛ. Інсулін проявляє антиліполітичний ефект, стимулює транспорт глюкози в адипоцитах та впливає на активність ЛПЛ [18, 19]. Симпатична нервова система відіграє важливу роль у регуляції ліполізу і мобілізації ліпідів в організмі людини. Катехоламіни впливають одночасно на адипоцити й васкуляризацію жирової тканини, контролюючи локальний кровообіг. Крім того, вони модулюють секрецію антиліполітичного гормона інсуліну.
З одного боку, висока ліполітична адренергічна активність адипоцитів вісцеральної жирової тканини і їх резистентність до антиліполітичної дії інсуліну зумовлюють надмірне надходження ВЖК у портальну систему печінки. З іншого боку, вісцеральна жирова тканина містить велику кількість імунореактивних клітин та преадипоцитів у стромально-судинній фракції, які за наявності ожиріння здатні секретувати різноманітні прозапальні цитокіни. Обидва фактори спричинюють розвиток гіперліпідемії, гіперінсулінемії, ІР і гіперглікемії [20].
У 1963 році Randle та співавт. на моделі ізольованого серця та діафрагми у щурів продемонстрували, що на фоні підвищеної концентрації ВЖК у крові спостерігається збільшення швидкості окислення жирів порівняно з вуглеводами. Вчені припустили, що конкуренція між даними метаболітами призводить до зниження споживання глюкози інсуліночутливими тканинами та відіграє важливу роль у розвитку ІР при ожирінні та ЦД2 [21]. Отримані результати підтверджуються в дослідженнях на здорових добровольцях, оскільки збільшення концентрації ВЖК в плазмі пригнічує утилізацію глюкози під час проведення гіпер- чи еуглікемічного гіперінсулінемічного клемпу (ЕГК) [22–24].
Подальші дослідження групи Шульмана підтвердили, що надлишок ВЖК знижує чутливість печінки й інших тканин до інсуліну через поставки конкурентного до глюкози субстрату, а також запропонували альтернативну гіпотезу, згідно з якою ІР формується внаслідок порушень у пострецепторній передачі інсулінового сигналу [25, 26]. При надлишку ВЖК у гепатоцитах формується надлишок ацил-КоА та його похідних типу церамідів і похідних фосфатидної кислоти. Похідні фосфатидної кислоти порушують роботу протеїн-кінази С і пострецепторне фосфорилювання тирозину в субстратах інсулінового рецептора IRS-I і IRS-II, особливо в носіїв їх мутантних алелів. Цераміди блокують роботу PkB/Akt сигнального шляху (рис. 1). Все це призводить до порушення активності транспортерів глюкози і зниження передачі сигналу від інсулінового рецептора. На тваринних моделях ожиріння фармакологічне чи генетичне блокування зазначених шляхів чи зменшення утворення церамідів не призводить до розвитку ІР після споживання збагаченої ліпідами дієти [27, 28].
/75/75.jpg)
З огляду на те, що в даний час роль ІР як ключової ланки розвитку МС є незаперечною, виникає необхідність у точному й відтворюваному методі для її вимірювання. На сучасному етапі виділяють непрямі й прямі методи кількісної оцінки дії інсуліну. Непрямі методи спрямовані на оцінку ефектів ендогенного інсуліну, і вони розраховуються за допомогою структурних математичних моделей на основі внутрішньовенного (постійна інфузія глюкози з модельною оцінкою — ПІГМО) і перорального ГТТ або визначення глюкози та інсуліну натще (з обчисленням індексів, у тому числі HOMA, QUICKI) [29, 30]. При проведенні прямих методів (екзогенні) здійснюють інфузію інсуліну й оцінюють його ефекти на метаболізм глюкози. Серед них інсуліновий тест толерантності, інсуліновий супресивний тест, еуглікемічний гіперінсулінемічний клемп [31].
Найбільш точним методом, золотим стандартом оцінки ІР як у хворих на ЦД2, так і здорових людей є ЕГК [32]. Перевагами ЕГК вважаються: можливість оцінки чутливості до інсуліну без ризику гіпоглікемії і подальшого викиду контрінсулярних гормонів без втручання ендогенного інсуліну і впливу різних рівнів гіперглікемії. Окрім вивчення метаболізму глюкози, використовуючи мічені ВЖК (або гліцерин) і амінокислоти, можна оцінювати вплив самого інсуліну на ліполіз і катаболізм білків. Окрім того, при поєднанні ЕГК з новітніми методами дослідження обміну речовин, такими як ПЕТ ([18F]-дезоксиглюкоза), катетеризація вен різних регіонів, непряма калориметрія і біопсія тканин, ядерно-магнітно-резонансна спектроскопія ([13C]-глюкоза), стає можливим вивчення складного механізму дії інсуліну, включаючи регуляцію поглинання, продукцію та метаболізм глюкози вибірково різними органами і тканинами (печінка, скелетні м’язи, жирова тканина, міокард), оцінити вплив інсуліну на накопичення глікогену, окислення субстратів і його стимулюючу дію на термогенез [31].
Недоліки методу ЕГК визначаються його складністю (потрібні два внутрішньовенних доступи, калібровані помпи й установка для швидкого і точного визначення рівня глюкози плазми). Наприкінці тесту, особливо при використанні високих доз інсуліну, рівень глікемії у пацієнта повинен деякий час моніторуватись через небезпеку розвитку гіпоглікемії [31]. Саме трудомісткість даного методу та його дорожнеча не дозволяють використовувати його в широкій клінічній практиці.
Найбільш простим і зручним для застосування в клінічній практиці методом оцінки ІР є гомеостатична модель оцінки (НОМА) ІР, яка була вперше описана в 1985 році [33]. У даній методиці для розрахунку використовується співвідношення концентрацій базального інсуліну чи С-пептиду й глюкози крові, що відображає баланс між ендогенною продукцією глюкози в печінці та секреції інсуліну bклітинами, що підтримується за принципом негативного оберненого зв’язку. Модель базується на двох основних припущеннях: по-перше, ступінь, при якому базальна концентрація глюкози зростає у відповідь на недостатність інсуліну, що відображає форму нормального секреторної відповіді інсуліну на глюкозу; по-друге, базальний рівень інсуліну прямо пропорційний ІР.
Графік зміни концентрацій інсуліну й глюкози плазми дозволяє передбачити співвідношення нестачі інсуліну та резистентності до нього:
НОМА-IR = імунореактивний інсулін (мкОД/мл) x глюкоза плазми натще / 22,5.
НОМА-бета = імунореактивний інсулін (мкОД/мл) x 20 / глюкоза плазми натще (ммоль/л) – 3,5.
Також можна проводити розрахунок даної моделі, скоригованої за допомогою спеціальної комп’ютерної програми — HOMA-2, у тому числі з використанням рівня С-пептиду замість імунореактивного інсуліну [34]. Крім того, в даному варіанті моделі HOMA додатково враховуються варіативні зміни ІР у печінці та периферичних тканинах, модель включає оцінку секреції проінсуліну та втрати глюкози нирками, що дозволяє використовувати її в пацієнтів із гіперглікемією. Комп’ютерна модель НОМА-2 використовується для визначення чутливості до інсуліну (% S) і бета-клітинної функції (% B). Результат наводиться у відсотках. За 100 % прийняті аналогічні показники, отримані у здорових людей молодого віку. Дана модель доступна за таким посиланням: www.OCDEM.ox.ac.uk
У пацієнтів із ЦД2 та здорових волонтерів встановлено наявність сильного позитивного кореляційного взаємозв’язку між ІР, оціненою за допомогою методу НОМА-IR та ЕГК (r > 0,85, p > 0,001) [33, 35], і відповідно ПІГМО (r > 0,75, p > 0,001) [36], що підтверджує можливість широкого використання даних розрахункових методів у клінічній практиці.
Жирова тканина є основним енергетичним депо в організмі. З усієї енергії, що надходить в організм з їжею, близько 75 % витрачається на підтримання основного обміну, близько 10–15 % від її кількості використовується на різні види фізичної активності і 10–15 % — на підтримку постійного термогенезу. Протягом тривалого періоду вважали, що жирова тканина є лише інертним енергетичним депо. Після виявлення ендокринної функції жирової тканини, і особливо після відкриття гіпоталамо-ліпоцитарної нейроендокринної осі, ліпоцентрична теорія патогенезу МС трансформувалася в ліпокінову теорію. З точки зору ліпокінової теорії основні складові МС формує не стільки субстратно-енергетична роль продуктів ліпоцитів, скільки інформаційний вплив на організм адипоцитарних сигнальних молекул [20].
На сьогодні жирова тканина — активний ендокринний орган, що виконує низку ендокринних, паракринних і автокринних функцій і в якому синтезується значна кількість гормонів і біологічно активних пептидів, до яких належать: лептин, пантофізин, резистин, фактор некрозу пухлини альфа (TNF-aльфа), адипонектин, вісфатин, внутрішньоадипоцитарні альтернативні білки (адипсин, С3, В), внутрішньоадипоцитарний білок 30 kD (ACRP30), білок, що стимулює ацетилювання (ASP), ЛПЛ, білок, що переносить ефіри холестерину, аполіпопротеїн Е (Apo E), ретинолзв’язувальний протеїн-4 (RBP-4), судинний ендотеліальний фактор росту (VEGF), інтерлейкін (ІЛ) 6, ангіотензиноген, інгібітор 1-го типу активатора плазміногену (PAI-1), трансформуючий фактор росту бета (TGF-бета), фактор росту гепатоцитів, інсуліноподібний фактор росту 1 (IGF-1), монобутирин, білки 1, 2 і 3-го типу, що роз’єднують окисне фосфорилювання, простациклін (PgI2), білки гострої фази (гаптоглобін, альфа-1-кислий глікопротеїн), білки позаклітинного матриксу (колаген 1, 3, 4 і 6-го типу, фібронектин; остеонектин; ламінін; матриксні металопротеїнази 2-го і 9-го типу), естрогени (р450-ароматаза конвертує андростендіон в естрон), 17-бета-гідроксистероїдна оксидоредуктаза, аgouti сигнальний білок та інші [37].
Лептин — гормон білкової природи з молекулярною масою 16 кДа, що секретується в основному в адипоцитах і в невеликій кількості в м’язах та плаценті. Відкритий у 1995 р. J.M. Friedman. Назва лептину походить від грецького слова leptos, що в перекладі означає «тонкий» [38].
Фізіологічна функція лептину полягає в запобіганні розвитку ожиріння в умовах надлишкового надходження їжі в організм. Зниження секреції лептину при голодуванні активує катаболізм та стимулює апетит. При надмірному надходженні їжі в організм лептин посилює термогенез шляхом активування енергоутворення в бурій жировій тканині за допомогою індукції експресії генів, відповідальних за синтез мітохондріальних білків 1, 2 і 3-го типу, що роз’єднують окисне фосфорилювання і регулюють швидкість термогенезу в організмі [37].
Рецептор лептину (Ob-R) був уперше ідентифікований Tartaglia et al. у 1995 р. [39]. Виділяють декілька сплайсингових варіантів OB-R: OB-Rа, OB-Rb, OB-Rc, OB-Rd, OB-Re і OB-Rf. Для всіх варіантів спільним є позаклітинний домен, до складу якого входить понад 800 амінокислот, трансмембранний домен із 34 амінокислот і варіабельний внутрішньоклітинний домен. Залежно від довжини внутрішньоклітинного домену ізоформи рецептора також поділяються на три класи: короткі, довгі та секретовані. До коротких зараховують OB-Rа, OB-Rc, OB-d і OB-Rf, цитоплазматичний домен яких містить 30–40 амінокислотних залишків [40]. Однак тільки довга ізоформа OB-Rb розглядається як функціональний рецептор з величиною внутрішньоклітинного домену в 300 амінокислотних залишків, який містить усі мотиви, необхідні для активації різних сигнальних шляхів. В OB-Re відсутній внутрішньоклітинний домен. Він являє собою розчинну форму рецептора, яка є альтернативним сплайсинговим варіантом або продуктом протеолітичної деградації мембранозв’язаних OB-R [41].
Лептинові рецептори розташовані в аркуатному та вентромедіальному ядрах гіпоталамуса, де локалізуються центри голоду, насичення і терморегуляції. В аркуатному ядрі ідентифіковано два типи клітин, один із яких відповідальний за утворення нейропептиду Y (NРY) і AgRP, які є пептидами, що стимулюють прийом їжі. Лептин знижує експресію генів зазначених білків. У клітинах другого типу лептин викликає підвищення експресії генів проопіомеланокортину (POMC) та амфетамінрегульованих транскриптів (CART), які кодують відповідні анорексигенні протеїни [37].
У людини вроджена недостатність лептину супроводжується ожирінням, гіперфагією і гіпогонадотропним гіпогонадизмом. Застосування екзогенного лептину зумовлює значне зниження апетиту, надлишкової маси тіла та ініціює розвиток пубертату.
Припущення про те, що недостатність секреції лептину в людини супроводжується ожирінням, не знаходить клінічного підтвердження. Рівень лептину в сироватці крові підвищується зі збільшенням ступеня ожиріння, тоді як доведена недостатність секреції лептину трапляється вкрай рідко. Ці дані дозволяють вважати, що при ожирінні відзначається резистентність до лептину. До етіологічних факторів лептинорезистентності зараховують: порушення синтезу білка, який зв’язує лептин у сироватці крові, патологію лептинових рецепторів, секрецію адипоцитами біологічно неактивних форм лептину, порушення транспорту лептину через гематоенцефалічний бар’єр, порушення на пострецепторному рівні передачі сигналу та гіперекспресію факторів, що забезпечують негативний зворотний зв’язок.
Встановлено, що лептин стимулює окислення жирних кислот, тим самим проявляючи протективний ефект проти ліпотоксичності. Проте тривалий час механізми, що забезпечують протидію проявам ліпотоксичності, не були відомі. Ситуація прояснилася після відкриття ролі лептину в селективному активуванні каталітичної a2-субодиниці аденозинмонофосфат-активованої протеїнкінази (AMPK) у скелетних м’язах. Активація AMPK підвищує bокислення жирних кислот шляхом блокування ефекту ацетил-КоА-карбоксилази (ACC). Після введення лептину експериментальним тваринам спостерігається підвищення рівня аденозинмонофосфату (АМФ) і активація AMPK вже через 15 хв. Така швидка відповідь обумовлена зв’язуванням лептину з Ob-Rb. Лептин також здатний викликати більш пізнє підвищення рівня АМФ шляхом активації aадренергічної системи в гіпоталамусі. Активацією AMPK принаймні частково можна пояснити вплив лептину на підвищене засвоювання глюкози [42, 43].
Інсулінозалежний ефект лептину характеризується дією на процеси глікогенолізу та глюконеогенезу та обумовлений активацією сигнального шляху PI3K, який регулюється широким спектром лігандів. Проте основним із них є інсулін. PI3K активує сигнальні каскади протеїнкінази В (Akt/PKB) і протеїнкінази С (PKC). Лептин діє через деякі компоненти сигнального каскаду інсуліну.
Фізіологічна концентрація лептину сироватки крові пригнічує другу фазу інсулінової секреції та експресію мРНК препроінсуліну. Ці ефекти оцінюють як один із проявів інгібіторної дії жирової тканини для уникнення надмірної стимуляції експресії препроінсулінового гена у відповідь на інкретини (глюкагоноподібний пептид-1) та глюкозу для запобігання розвитку гіперінсулінемії [44].
Лептин вважається прозапальним цитокіном та має подібну структуру до інших прозапальних цитокінів — ІЛ-6, ІЛ-12 і гранулоцитарного фактора. У мишей із мутацією в гені, що кодує лептин (ob/ob), або гені, що кодує рецептор лептину (db/db), які використовуються в багатьох дослідженнях як експериментальні моделі ожиріння, спостерігаються різного роду дефекти клітинного та гуморального імунітету [45].
У моноцитах і макрофагах лептин стимулює синтез прозапальних цитокінів — TNF-aльфа, ІЛ-6 і ІЛ-12. Індукована лептином продукція TNF-aльфа в мишачих перитонеальних макрофагах пригнічується глобулярним адипонектином через блокування фосфорилювання кіназ родини мітогенактивованих протеїнкіназ (MAPK — ERK1) [46]. У клітинах Купфера стимульований ліпополісахаридом лептин посилює продукцію TNF-aльфа, активуючи р38- і JNK/МАРК-сигнальні шляхи [47].
Lord та співавт., вивчаючи Т-клітинну проліферацію на мишах, продемонстрували, що лептин підвищує продукцію цитокінів Т-хелперами 1-го типу (TH1) — ІЛ-2 і інтерферону гамма (IFN-гамма), і пригнічує T-хелперами 2-го типу (TН2) — ІЛ-4, що відіграє важливу роль у патогенезі автоімунних захворювань [45]. Дефіцит лептину має протекторну дію, зменшуючи продукцію прозапальних цитокінів TH1 і переключаючи фенотип імунної відповіді на TН2 [48]. Продемонстровано, що миші лінії ob/ob резистентні до експериментально індукованого автоімунного енцефаломієліту [49].
Використання лептину як терапевтичного агента обмежене через виражену лептинорезистентність у більшості осіб, які страждають від ожиріння. На сьогодні терапія лептином успішно використовується тільки у хворих із генетичним дефіцитом лептину або ліподистрофією [50].
З урахуванням значення лептину у регуляції енергетичного обміну та харчової поведінки актуальним є дослідження його молекулярних механізмів дії для створення ефективних терапевтичних засобів лікування ожиріння та супутніх захворювань [51].
Адипонектин — колагеноподібний білковий гормон масою 30 кДа, який експресується переважно в жировій тканині, бере участь у регуляції катаболізму жирних кислот, чутливості до інсуліну, рівня глюкози в крові та інших процесів.
Повна молекула адипонектину представлена у вигляді трьох олігомерних комплексів: тримерів — LMW-форма (low molecular weight), гексамерів — MMW-форма (medium molecular weight), а також 12- та 18-мерів — HMW-форма (high molecular weight) [52]. Мономерів адипонектину в крові не виявлено, полімеризація білка відбувається в середині адипоцитів.
Рівень адипонектину в плазмі вірогідно знижений при вісцеральному ожирінні та патологічних станах, для яких характерна ІР: ЦД2, МС, неалкогольна жирова хвороба печінки, атеросклероз [53, 54]. Всі олігомерні форми адипонекину присутні в крові. Групою вчених висловлено припущення, що співвідношення (а не абсолютна кількість) HMW/LMW форм адипонектину в сироватці крові має вирішальне значення у визначенні чутливості до інсуліну периферичних тканин [55]. Помірна втрата ваги призводить до відносного збільшення співвідношення HMW/MMW та зниження абсолютної кількості LMW-форми адипонектину в сироватці крові [56].
Існують два типи рецепторів, які специфічно взаємодіють з адипонектином: AdipoR1 і AdipoR2. AdipoR1 (375 амінокислот, молекулярна маса 43 кДa) має високу афінність до глобулярного адипонектину і низьку афінність до олігомерних форм гормона. Рецептор у великій кількості експресується в скелетних м’язах, меншою мірою — у мозку, серці, нирках, печінці, плаценті, легенях, селезінці, лейкоцитах [57].
AdipoR2 (386 амінокислот, 44 кДа) має середню афінність до обох форм адипонектину. Амінокислотна послідовність AdipoR2 на 66,7 % аналогічна послідовності AdipoR1 [58]. AdipoR2 у великій кількості експресується в скелетних м’язах, печінці і плаценті, слабо в мозку, серці, селезінці, нирках, лейкоцитах і легенях.
Адипонектин кодується геном АPM1, що розташований у хромосомному регіоні 3q27. Даний регіон ідентифікований як локус, асоційований із розвитком ЦД2 та МС, а ген АPM1 виступає в ролі гена-кандидата. Декілька SNP (Single-nucleotide polymorphisms) у промоторі гена АPM1 асоційовані з ризиком розвитку ЦД2 в японській популяції та у кавказців із Франції і Скандинавії [60–62].
Дві групи вчених незалежно одна від одної досліджували наслідки делеції гена APM1 на чутливість до інсуліну [63]. Групи Kadowaki та Matsuzawa виявили, що у мишей із нокаутованим геном адипонектину спостерігається ІР, хоча були деякі незначні відмінності під час експерименту у двох груп. Kadowaki показав, що у мишей із генотипом адпонектин+/– розвивається ІР та ПТГ на стандартній дієті, які прогресують у мишей адпонектин–/– дозозалежним чином [64]. Група Matsuzawa спостерігала виражену ІР у поєднанні з дефектами в пострецепторній передачі інсулінового сигналу тільки після вигодовування мишей із нокаутованим геном адипонектину (–/–) дієтою з високим вмістом жирів [65].
Scherer вивів лінію трансгенних мишей із триразовим підвищенням рівня адипонектину в сироватці крові. Для даної моделі гіперадипонектинемії характерно підвищення чутливості периферичних тканин до інсуліну за рахунок покращення вуглеводного та ліпідного метаболізму, пов’язаного з підвищенням активації AMPK в печінці й експресії PPAR-гамма у вісцеральній жировій тканині. Дані тварини стійкі до розвитку ІР, індукованої дієтою з високим вмістом жирів [66].
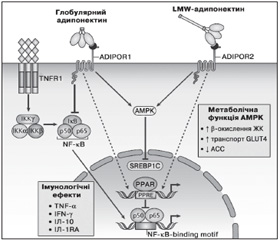
AMPK є сенсором енергетичного статусу клітини і відіграє ключову роль у забезпеченні системного енергетичного балансу за рахунок регулювання прийому їжі, маси тіла, метаболізму глюкози та ліпідів (рис. 2). Олігомерні форми адипонектину стимулюють фосфорилювання та активацію AMPK у печінці, у той час як глобулярний адипонектин проявляє даний ефект як у скелетних м’язах, так і тканині печінки. Адипонектин знижує рівень глюкози в крові за рахунок активації АМРК і інгібування ACC. АМРК збільшує продукцію енергії (споживання глюкози і жирних кислот) та інгібує енерговитратні реакції (глюконеогенез і синтез жирних кислот) [67].
/78/78.jpg)
Адипонектин стимулює синтез важливих прозапальних цитокінів, таких як ІЛ-10 та ІЛ-1RA (ІЛ-1 антагоніст рецептора), у моноцитах, макрофагах, дендритних клітинах, а також пригнічує утворення інтерферону гамма (IFN-гамма) у ЛПС-стимульованих макрофагах [68]. HMW, але не LMW і MMW олігомерні форми адипонектину захищають клітини ендотелію судин від апоптозу. При цьому вплив адипонектину на моноцити і макрофаги двоякий. Тример адипонектину пригнічує секрецію ІЛ-6 та IFN-гамма, що виділяються макрофагами, і стимулює виділення протизапальних цитокінів ІЛ-10 і ІЛ-1RA. На противагу цьому HMW-адипонектин збільшує виділення ІЛ-10 із моноцитів [69].
Вісфатин — поліпептид із молекулярною масою 52 кДа, що складається з 491 амінокислотного залишку. У 2005 р. Fukuhara et al. [70] відкрив вісфатин — новий адипоцитокін, що експресується переважно у вісцеральній жировій тканині. Даний гормон має інсуліноміметичну дію, зв’язуючись із рецепторами інсуліну в місцях, що відрізняються від сайтів зв’язування інсуліну, і, як наслідок, покращує толерантність до глюкози та відіграє важливу роль у патогенезі ожиріння, ІР та ЦД2 [71]. Проте в 2007 р. група Fukuhara et al. зазначила, що описаний ними адипоцитокін був ідентифікований раніше іншими лабораторіями як PBEF (pre-cell colony enhancing factor) — цитокін, що експресується в лімфоцитах та Nampt (нікотинамід фосфорибозилтрансфераза) — ключовим ферментом біосинтезу нікотинамідаденіндинуклеотиду (НАД) в організмі ссавців [70].
Вісфатин зв’язується з рецептором інсуліну і стимулює фосфорилювання субстратів IRS-1 і IRS-2 з подальшою активацією PI3K-, Akt/PKB-, МАРК-сигнальних шляхів. Вперше потенційна роль вісфатину як інсуліноміметика була продемонстрована Fukuhara et al., і ними ж проведена детальна характеристика даного адипоцитокіну. У мишей лінії C57BL/6J та KK-Aу (експериментальна модель ЦД2) у відповідь на введення рекомбінантного вісфатину спостерігався дозозалежний цукрознижувальний ефект. Хронічна експресія гормона за допомогою аденовірусного вектора у мишей C57BL/6J і KK-Aу призводила до значного зниження концентрації глюкози в плазмі. У дослідженні in vitro інсуліноміметичний ефект спостерігався при концентрації, у 10 разів нижчій порівняно з інсуліном [71].
У мишей із нокаутованим геном вісфатину–/– спостерігалась летальність під час ембріогенезу у зв’язку з порушенням біосинтезу НАД. У мишей із гетерозиготним генотипом (вісфатин+/–) спостерігається на 33 % нижча концентрація адипоцитокіну порівняно з мишами дикого типу та помірне підвищення рівня глюкози в плазмі крові натще і в постпрандіальний період, а також значно вищий рівень глікемії під час ГТТ порівняно з контрольною групою. Порівняно з інсуліном вісфатин проявляє аналогічну афінність до інсулінового рецептора. Вивчаючи конкурентне зв’язування інсуліну та вісфатину/PBEF/Nampt з інсуліновим рецептором, вчені виявили, що адипоцитокін зв’язується з іншими від інсуліну сайтами рецептора і стимулює його відмінним від інсуліну шляхом [71].
Вісфатин бере участь у регуляції запальних процесів та виступає в ролі імуномодулятора. Nampt вперше була ідентифікована у людей у лімфоцитах периферичної крові й отримала назву PBEF (pre-B-cell colony enhancing factor) [72]. Введення рекомбінантного вісфатину стимулює продукцію прозапальних — ІЛ-1b, TNF-aльфа та ІЛ-6 і протизапальних цитокінів — ІЛ-10, ІЛ-1RA моноцитами, а також підвищує експресію поверхневих костимуляційних молекул CD54, CD40 і CD80, необхідних для активації Т-лімфоцитів [73].
Berndt et al. на популяції з 189 чоловік показали, що концентрація вісфатину в плазмі крові та експресія його мРНК у вісцеральній жировій тканині позитивно корелює з ІМТ і процентним вмістом жиру в тілі [74].
Dogru et al. рандомізували 22 пацієнти з ЦД2 без попереднього лікування, 18 пацієнтів із ПТГ і 40 осіб сформували контрольну групу. У даному дослідженні не спостерігалось кореляції між концентрацією вісфатину та ІМТ, АТ, адипонектином, С-реактивним протеїном, інсуліном, глюкозою, ліпідами та HOMA-IR. Проте в групі з ЦД2 відзначався значно вищий рівень гормона порівняно з контрольною групою. У пацієнтів з ПТГ та ЦД2 суттєвих відмінностей у концентрації вісфатину не спостерігалось [75].
TNF-aльфа, прозапальний цитокін із молекулярною масою 17 кДа, синтезується моноцитами/макрофагами, нейтрофілами, Т-лімфоцитами, а також клітинами ендотелію та жирової тканини. У печінці TNF-aльфа продукується клітинами Купфера і в значно меншій кількості гепатоцитами [76, 77]. Його дія опосередковується двома типами рецепторів — TNFR1 (р55) та TNFR2 (р75). На моделі генетично детермінованого ожиріння (ob/ob) вчені продемонстрували протекторний ефект від виключення генів рецепторів TNF-aльфа (р55 –/– р75 –/–) на розвиток ІР порівняно з тваринами з функціонуючими рецепторами (р55 +/+ р75 +/+). У подальшому при селективному виключенні окремих генів виявилось, що ключову роль відіграє ген TNFR1 [78].
Hotamisligil et al. уперше продемонстрували взаємозв’язок між експресією TNF-aльфа та інсулінорезистентністю у жінок з ожирінням та неалкогольним стеатогепатитом. Вчені виявили, що жирова тканина у хворих з ожирінням є важливим джерелом прозапальних цитокінів, зокрема TNF-aльфа, який індукує запалення та ІР [79].
На моделях експериментального ожиріння, індукованого висококалорійною дієтою (5286 ккал/кг–1) та дієтою з підвищеним умістом жирів (50 % жирів від загального калоражу), у мишей із TNF–a +/+ та нокаутованим геном TNF-aльфа (TNF-aльфа –/–) спостерігалась надмірна маса тіла порівняно з контролем, проте вірогідної різниці між двома експериментальними групами не відзначалося. Проте, незважаючи на аналогічну динаміку набирання маси тіла, у мишей із виключеним геном TNF-aльфа спостерігалась підвищена чутливість периферичних тканин до інсуліну [80].
Одну з ключових ролей у розвитку TNF-aльфа-індукованої ІР відіграє активація JNK1 (c-Jun amino-terminal kinase). Hirosumi et al. вперше продемонстрували на експериментальних моделях індукованого дієтою та генетично детермінованого (ob/ob) ожиріння підвищення активності JNK1 в печінці, м’язовій та жировій тканинах. Блокада гена JNK1 призводила до зменшення ожиріння, зниження рівня глікемії, резистину та ІР, підвищення сироваткового рівня адипонектину на обох експериментальних моделях ожиріння. Обробка культури гепатоцитів TNF-aльфа призводила до розвитку в них ІР, яка нівелювалась після введення інгібітору JNK1. JNK1 індукує ІР шляхом підвищеного фосфорилювання залишку серину в 307-му положенні в субстраті інсулінового рецептора — IRS1, тим самим блокуючи його біологічну активність [81].
Іншим посередником у TNF-aльфа-індукованій ІР є IKK-b [82], що є структурною субодиницею IkB кінази (ІКК) — ферменту, який каталізує фосфорилювання інгібіторних протеїнів kB. Ядерний фактор kB (NF-kB) у неактивному стані, локалізований у цитоплазмі, перебуває в комплексі з інгібіторними протеїнами kB (IkB), переважно IkBa. При фосфорилюванні IkBa фактор транскрипції NF-kB вивільняється із зв’язку з IkB, мігрує в ядро клітини і стимулює транскрипцію багатьох прозапальних генів, що кодують синтез адипокінів та цитокінів (ІЛ-6, TNF-aльфа) [83] та порушує трансдукцію інсулінового сигналу шляхом фосфорилювання залишків серину в IRS-1 [84]. На моделях трансгенних мишей безперервна експресія IKK-b на низькому рівні в гепатоцитах призводила до активації NF-kB з подальшим розвитком помірно вираженої ІР [85]. У хворих на ЦД2 блокування високими дозами аспірину (7 г) IKK-b призводило до покращення чутливості до інсуліну периферичними тканинами [86].
Ожиріння характеризується підвищеною експресією в жировій тканині цитокінів родини ІЛ-1, серед яких частина має виражену прозапальну активність — ІЛ-1a, ІЛ-1b та ІЛ-18, а інші є протизапальними медіаторами — антагоніст рецептора IЛ-1 (ІЛ-1Ra) та ІЛ-37 [87]. На експериментальних моделях генетично детермінованого та індукованого дієтою з високим вмістом жирів ожиріння продемонстровано підвищення активності ІЛ-1b у піддослідних тварин. На думку Moschen et al., за умови патологічного ожиріння саме жирова тканина є основним джерелом ІЛ-1b, оскільки його експресія значно вища в підшкірній/вісцеральній жировій тканині порівняно з печінкою [88].
Цитокіни — члени родини ІЛ-1 — беруть участь у метаболізмі глюкози та в розвитку ІР [89]. Надмірне виділення ІЛ-1b жировою тканиною у мишей із генетично детермінованим ожирінням та ІР контролює чутливість гепатоцитів до інсуліну [90]. ІЛ-1b на транскрипційному рівні зменшує експресію субстрату інсулінового рецептора IRS-1 через ERK-залежні та незалежні механізми, тим самим провокуючи розвиток ІР [91]. Введення мишам із дієт-індукованим ожирінням нейтралізуючих анти-ІЛ-1b-антитіл XOMA 052 призводило до підвищення чутливості периферичних тканин до інсуліну й покращення бета-клітинної функції [92]. Лікування хворих на ЦД2 рекомбінантним людським ІЛ-1Ra покращує глікемічний контроль [93].
Резистин був ідентифікований у 2001 році як гормон жирової тканини, експресія якого пригнічується після введення агоністів PPAR-гамма [94]. В організмі тварин адипоцитокін переважно синтезується преадипоцитами та складається зі 114 амінокислотних залишків. У той час як у мишей експресія резистину відбувається виключно в білій жировій тканині, у людини резистин в основному секретується циркулюючими моноцитами [95] і тільки на 64 % гомологічний резистину мишей [96].
В експериментальних тварин із генетично детермінованим ожирінням і діабетом (моделі ob/ob і db/db) спостерігається підвищення концентрації резистину в сироватці. Введення резистину мишам призводить до розвитку ПТГ, а в мишей із дієт-індукованим ожирінням на фоні ін’єкцій моноклональних антитіл до резистину відзначалось зменшення ІР та зниження гіперглікемії [97–99]. Інфузія резистину в умовах нормоглікемії і гіперінсулінемії індукує печінкову, але не периферичну резистентність до інсуліну у щурів і, таким чином, відповідальна за підвищення швидкості утворення глюкози печінкою [97]. У мишей із нокаутованим геном резистину спостерігається покращення гомеостазу глюкози, що обумовлено підвищенням активності AMPK і зниженням експресії генів основних ферментів глюконеогенезу в печінці [100]. Крім того, резистин індукує експресію SOCS-3, транскрипційного фактора, який є негативний регулятором передачі інсулінового сигналу [101].
Всі ці дані свідчать про те, що збільшення секреції резистину у тварин призводить до ожиріння і ІР, що може бути сполучною ланкою між ожирінням і ЦД. Роль резистину в організмі людини є менш визначеною. За даними епідеміологічних досліджень не вдалось виявити кореляційних взаємозв’язків між вмістом резистину в крові і розвитком ожиріння та ІР [102, 103].
ІЛ-6 — прозапальний цитокін, синтезується активованими моноцитами, менше фібробластами, ендотелієм при запаленні, гіпоксії, дії бактеріальних ендотоксинів [104]. До 30 % циркулюючого ІЛ-6 синтезується адипоцитами [105], причому у вісцеральній жировій тканині у 2–3 рази вище порівняно з підшкірною [106].
Відомо, що ожиріння, МС, ЦД2 супроводжуються запаленням жирової тканини. При даних патологічних станах секреція ІЛ-6 підвищується і його концентрація в крові зростає, досягаючи значень 100 пг/мл [107, 108] порівняно з референтним значенням в 1–2 пг/мл у здорових добровольців. Ступінь підвищення рівня ІЛ-6 незалежно асоційований із вираженістю ІР [109] та є індикатором збільшення маси жирової тканини в організмі [110].
На культурі адипоцитів продемонстровано, що тривала експозиція з ІЛ-6 призводить до пригнічення експресії генів IRS-1 та GLUT-4, що проявляється зменшенням інсулінзалежного засвоєння глюкози [111]. Крім того, за даних умов ІЛ-6 зменшує експресію гена адипонектину та активує експресію низки цитокінів, у тому числі TNF-aльфа [111, 112].
У гепатоцитах ІЛ-6 сприяє вивільненню глюкози, стимулює глікогеноліз за рахунок активації глікогенфосфорилази і гальмування інсулінзалежного синтезу глікогену [113]. Молекулярний механізм інгібуючого впливу ІЛ-6 на дію інсуліну в печінці полягає в синтезі SOSC-3, який, зв’язуючись із IRS-1, блокує передачу інсулінового сигналу від його рецептора [114].
У мишей із генетичним нокаутом гена ІЛ-6 –/– спостерігається ожиріння, збільшення на 50–60 % кількості жирової тканини, гіперглікемія та порушується засвоєння глюкози, що свідчить про розвиток ІР на системному рівні. Також такі експериментальні тварини не здатні до тривалих фізичних навантажень, а засвоєння кисню під час тренування у них нижче, ніж у контролі [115].
Суперечливі результати отримані також при вивченні дії ІЛ-6 на чутливість тканин до інсуліну на системному рівні. При введенні ІЛ-6 людині або гризунам відзначено як поліпшення, так і відсутність ефекту або ж погіршення дії інсуліну на рівні цілого організму [113, 116, 117]. Однією з причин протилежних результатів дії ІЛ-6 на інсуліновий сигнальний шлях можуть бути особливості ефектів цитокіну в м’язовій тканині. Якщо в печінкових і жирових клітинах ІЛ-6 сприяє розвитку ІР, то в м’язових він, навпаки, посилює ефекти інсуліну [118]. Причини дуалістичних ефектів ІЛ-6 на дію інсуліну в різних тканинах організму до кінця не з’ясовані. Проте, на думку Шварца, певне значення може мати часова характеристика: підвищується секреція ІЛ-6 транзиторно, як при фізичній активності, або перманентно, як при хронічній системній запальній відповіді, що типово для ожиріння, МС, ЦД2. Короткочасне підвищення концентрації ІЛ-6 у крові і тканинах служить сигналом енергетичного дефіциту й посилює дію інсуліну в м’язових клітинах і пригнічує його в тканинах, що постачають енергетичні субстанції: печінка і жирова тканина. Мабуть, у цьому полягають причини різних, часом протилежних, ефектів ІЛ-6 на обмінні процеси, особливо на дію інсуліну в тканинах [119].
1. Reaven G.M. Role of insulin resistance in human disease / G.M. Reaven // Diabetes. — 1988. — V. 37. — P. 1595–1607.
2. Kaplan N.M. The deadly quartet: upper–body obesity, glucose intolerance, hypertriglyceridemia and hypertension / N.M. Kaplan // Arch. Intern. Med. — 1989. — V. 149. — P. 1514–1520.
3. Henefeld M. Das metabolische Syndrome. Deutsch / M. Henefeld, W. Leonhardt // Ges. Wes. — 1980. — V. 36. — P. 545–551.
4. DeFronzo R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture, 2009 / R.A. DeFronzo // Diabetologia. — Vol. 53, № 7. — Р. 1270–1287.
5. Body mass index and mortality in a prospective cohort of US adults / E.E. Calle, M.J. Thun, J. M. Petrelli [et al.] // N. Engl. J. Med. — 1999. — Vol. 341. — P. 1097–1105.
6. The disease burden associated with overweight and obesity / A. Must, J. Spadano, E.H. Coakley // JAMA. — 1999. — Vol. 282. — P. 1523–1529.
7. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR) / E. Ferrannini, A. Natali, P. Bell [et al.] // J. Clin. Invest. — 1997. — Vol. 100. — P. 1166–1173.
8. What are the physical characteristics associated with a normal metabolic profile despite a high level of obesity in postmenopausal women? / M. Brochu, A. Tchernof, I.J. Dionne [et al.] // J. Clin. Endocrinol. Metab. — 2001. — Vol. 86. — P. 1020–1025.
9. The metabolically healthy but obese individual presents a favorable inflammation profile / A.D. Karelis, M. Faraj, J.P. Bastard [et al.] // J. Clin. Endocrinol. Metab. — 2005. — Vol. 90. — P. 4145–4150.
10. Karelis A.D. Can we identify metabolically healthy but obese individuals (MHO)? / A.D. Karelis, M. Brochu, R. Rabasa–Lhoret // Diabetes Metab. 2004. — Vol. 30. — P. 569–572.
11. Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease / J.B. Meigs, P.W. Wilson, C.S. Fox // J. Clin. Endocrinol. Metab. — 2006. — Vol. 91. — P. 2906–2912.
12. Impaired fasting glucose and impaired glucose tolerance: implication for care / D.M. Narthan, M.B. Davidson, R.A. De Fronzo [et al.] // Diabetes Care. — 2007. — Vol. 30. — P. 753–759.
13. Identification of individuals with insulin resistance using routine measurements / S.E. Stern, K. Williams, E. Ferranini [et al.] // Diabetes. — 2005. — Vol. 54. — P. 333–339.
14. The International Expert Committee. International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes // Diabetes Care. — 2009. — Vol. 32, № 7. — P. 1327–1334.
15. American Diabetes Associations. Diagnosis and classification of diabetes mellitus // Diabetes Care. — 2010. — S. suppl. 1. — S. 62–69.
16. Строев Ю.И., Цой М.В., Чурилов Л.П., Шишкин А.Н. Классические и современные представления о метаболическом синдроме. Ч. 2. Патогенез // Вестн. С.–Петерб. ун–та. — Сер. 11, Вып. 4. — 2007. — С. 3–15.
17. Bjorntorp P. «Portal» adipose tissue as the generator of risk factors for cardiovascular disease and diabetes / P. Bjorntorp // Arteriosclerosis. — 1990. — Vol. 10. — P. 493–496.
18. Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues from obese women and men / S.K. Fried, C.D. Russsell, N.L. Grauso, R.E. Brolin // J. Clin. Invest. — 1991. — Vol. 92. — P. 2191–2198.
19. Glucocorticoids down–regulate glucose uptake capacity and insulin–signalling proteins in omental but not subcutaneous adipocytes / M. Lundgren, J. Buren, T. Ruge [et al.] // J. Clin. Endocrinol. Metab. — 2004. — Vol. 89. — P. 2989–2997.
20. Малижев В.О. Дисфункція жирової тканини як вирішальний чинник розвитку цукрового діабету 2 типу // Здоров’я України. — 2007. — № 10/1.
21. The glucose fatty–acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus / P.J. Randle, P.B. Garland, C.N. Hales, E.A. Newsholme // Lancet. — 1963. — Vol. 13, № 1. — Р. 785–789.
22. Effect of long chain triglyceride infusion on glucose metabolism in man / D. Thiebaud, R. A. DeFronzo, E. Jacot [et al.] // Metabolism. — 1982. — Vol. 31, № 11. — Р. 1128–1136.
23. Effect of fatty acids on glucose production and utilization in man / E. Ferrannini, E.J. Barrett, S. Bevilacqua, R.A. DeFronzo // Journal of Clinical Investigation. — 1983. — Vol. 72, № 5. — Р. 1737–1747.
24. Interaction between glucose and free fatty acid metabolism in human skeletal muscle / D.E. Kelley, M. Mokan, J.A. Simoneau, L.J. Mandarino // Journal of Clinical Investigation. — 1993. — Vol. 92, № 1. — Р. 91–98.
25. Effects of free fatty acids on glucose transport and IRS–1–associated phosphatidylinositol 3–kinase activity / A. Dresner, D. Laurent, M. Marcucci [et al.] // Journal of Clinical Investigation. — 1999. — Vol. 103, № 2. — Р. 253–259.
26. Shulman G.I. Cellular mechanisms of insulin resistance / G.I. Shulman // Journal of Clinical Investigation. — 2000. — Vol. 106, № 2. — Р. 171–176.
27. Inhibition of ceramide synthesis ameliorates glucocorticoid–, saturated–fat–, and obesity–induced insulin resistance / W.L. Holland, J.T. Brozinick, L.P. Wang [et al.] // Cell Metab. — 2008. — Vol. 5. — P. 167–179.
28. Local and systemic insulin resistance resulting from hepatic activation of IKK–beta and NF–kappaB / D. Cai, M. Yuan, D.F. Frantz // Natю Med. — 2005. — Vol. 11. — P. 183–190.
29. Groop L.C. Insulin resistance and insulin deficiency in the pathogenesis of type 2 (non–insulin–dependent) diabetes mellitus: errors of metabolism or of methods? / L.C. Groop, E. Widйn, E. Ferrannini // Diabetologia. — 1993. — Vol. 36, № 12. — P. 1326–1331.
30. Майоров А.Ю. Методы количественной оценки инсулинорезистентности / А.Ю. Майоров, К.А. Урбанова, Г.Р. Галстян // Ожирение и метаболизм. — 2009. — № 2. — С. 19–23.
31. Алишева Е.К. Методы ранней диагностики инсулинорезистентности / Е.К. Алишева, Е.И. Красильникова, Е.В. Шляхто // Артериальная гипертензия. — 2002. — Т. 8, № 1. — С. 29–34.
32. DeFronzo R.A. Glucose clamp technique: a method for quantifying insulin secretion and resistance / R.A. DeFronzo, J.D. Tobin, R. Andres // American Journal of Physiology. — 1979. — Vol. 237, № 3. — P. 214–223.
33. Homeostasis model assessment: insulin resistance and b–cell function from fasting plasma glucose and insulin concentrations in man / D.R. Matthews, J.P. Hosker, A.S. Rudenski [et al.] // Diabetologia. — 1985. — Vol. 28, № 7. — P. 412–419.
34. Wallace T.M. Use and abuse of HOMA modeling / T.M. Wallace, J.C. Levy, D.R. Matthews // Diabetes Care. — 2004. — Vol. 27, № 6. — P. 1487–1495.
35. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity / E. Bonora, G. Targher, M. Alberichie [et al.] // Diabetes Care. — 2000. — Vol. 23. — P. 57–63.
36. Comparison of several insulin sensitivity indices derived from basal plasma insulin and glucose levels with minimal model indices / D.A. Garcia–Estevez, D. Araujo–Vilar, G. Fiestras–Janeiro // Horm. Metab. Res. — 2003. — Vol. 35. — P. 13–17.
37. Инсулиновая резистентность и роль гормонов жировой ткани в развитии сахарного диабета: пособие для врачей / Дедов И.И., Балаболкин М.И., Мамаева Г.Г. [и др.] // М., 2005. — 88 с.
38. Friedman J.M. Leptin at 14 y of age: an ongoing story / J.M. Friedman // Am. J. Clin. Nutr. — 2009. — Vol. 89(Suppl.). — P. 973–979.
39. Identification and expression cloning of a leptin receptor, OB–R / L.A. Tartaglia, M. Dembski, X. Weng [et al.] // Cell. — 1995. — Vol. 83. — P. 1263–1271.
40. A novel leptin receptor isoform in rat / M.Y. Wang, Y. Zhou, C.B. Newgard, R.H. Unger // FEBS Lett. — 1998. — Vol. 392. — P. 87–90.
41. Abnormal splicing of the leptin receptor in diabetic mice / G.H. Lee, R. Proenca, J.M. Montez [et al.] // Nature. — 1996. — Vol. 379. — P. 632–635.
42. Minokoshi Y. Role of AMP–activated protein kinase in leptin–induced fatty acid oxidation in muscle / Y. Minokoshi, B.B. Kahn // Biochem. Soc. Trans. — 2003. — Vol. 31. — P. 196–201.
43. AMPK expression and phosphorylation are increased in rodent muscle after chronic leptin treatment / G.R. Steinberg, J.W.E. Rush, D.J. Dyck // Am. J. Physiol. Endocrinol. Metab. — 2003. — Vol. 284. — P. 648–654.
44. Mohammed J. Adipokines and pathogenesis of non–alcoholic fatty liver disease / J. Mohammed, Y. Zobair // AnCha Baranova. — 2008. — 192 p.
45. Leptin modulates the T–cell immune response and reverses starvation–induced immunosuppression / G.M. Lord, G. Matarese, J.K. Howard [et al.] // Nature. — 1998. — Vol. 394, № 6696. — P. 897–901.
46. Globular adiponectin decreases leptin–induced tumor necrosis factor– expression by murine macrophages: involvement of cAMP–PKA and MAPK pathways / T. Zhao, M. Hou, M. Xia [et al.] // Cell Immunol. — 2005. — Vol. 238, № 1. — P. 19–30.
47. Leptin enhances TNF– production via p38 and JNK MAPK in LPS–stimulated Kupffer cells / J. Shen, I. Sakaida, K. Uchida [et al.] // Life Sci. — 2005. — Vol. 77. — P. 1502–1515.
48. Leptin–deficient (ob/ob) mice are protected from T cell–mediated hepatotoxicity: role of tumor necrosis factor–б and ІЛ–18 / R. Faggioni, J. Jones–Carson, D.A. Reed [et al.] // Proc. Natl Acad. Sci. USA. — 2000. — Vol. 97, № 5. — P. 2367–2372.
49. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis / G. Matarese, A. Di Giacomo, V. Sanna [et al.] // J. Immunol. — 2001. — Vol. 166, № 10. — P. 5909–5916.
50. Gorden P. The clinical uses of leptin / P. Gorden, O. Gavrilova // Curr. Opin. Pharmacol. — 2003. — Vol. 3. — P. 655–659.
51. Кобиляк Н.М. Патофізіологічна роль лептину у розвитку ожиріння та супутніх захворювань / Н.М. Кобиляк, М.М. Кондро, О.В. Вірченко, Т.М. Фалалєєва // Експериментальна та клінічна фізіологія і біохімія. — 2013. — № 3 (63). — С. 55–63.
52. Structure–function studies of the adipocyte–secreted hormone Acrp30/adiponectin. Implications for metabolic regulation and bioactivity / U.B. Pajvani, X. Du, T.P. Combs [et al] // J. Biol. Chem. — 2003. — Vol. 278, № 11. — P. 9073–9085.
53. Патофізіологічна роль адипонектину в розвитку ожиріння та супутніх захворювань / Н.М. Кобиляк, Г.П. Михальчишин, О.А. Савченюк, Т.М. Фалалєєва // Світ медицини та біології. — 2013. — № 3(40), ч. 2. — С. 81–87.
54. Михальчишин Г.П. Гіпоадипонектинемія у хворих на цукровий діабет типу 2 з неалкогольною жировою хворобою печінки / Г.П. Михальчишин, П.М. Боднар, Н.М. Кобиляк // Ендокринологія. — 2013. — Т. 18, № 2. — С. 18–25.
55. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione–mediated improvement in insulin sensitivity / U.B. Pajvani, M. Hawkins, T.P. Combs [et al.] // J. Biol. Chem. — 2004. — Vol. 279, № 13. — Vol. 12152–12162.
56. Changes of adiponectin oligomer composition by moderate weight reduction / T. Bobbert, H. Rochlitz, U. Wegewitz [et al.] // Diabetes. — 2005. — Vol. 54, № 5. — P. 2712–2719.
57. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects / T. Yamauchi, J. Kamon, Y. Ito [et al.] // Nature. — 2003. — Vol. 423, № 6941. — P. 762–769.
58. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome / T. Kadowaki, N. Yamauchi, N. Kubota [et al.] // J. Clin. Invest. — 2006. — Vol. 116, № 7. — P. 1784–1792.
59. Tilg H. Adipocytokines: mediators linking adipose tissue, inflammation and immunity / H. Tilg, A.R. Moschen // Nat. Rev. Immunol. — 2006. — Vol. 6, № 10. — P. 772–783.
60. Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population / K. Hara, P. Boutin, Y. Mori [et al.] // Diabetes. — 2002. — Vol. 51. — P. 536–540.
61. Single–nucleotide polymorphism haplotypes in the both proximal promoter and exon 3 of the APM1 gene modulate adipocytesecreted adiponectin hormone levels and contribute to the genetic risk for type 2 diabetes in French Caucasians / F. Vasseur, N. Helbecque, C. Dina [et al.] // Hum. Mol. Genet. — 2002. — Vol. 11. — P. 2607–2614.
62. Single nucleotide polymorphisms in the proximal promoter region of the adiponectin (APM1) gene are associated with type 2 diabetes in Swedish Caucasians / H.F. Gu, A. Abulaiti, C.G. Ostenson [et al.] // Diabetes. — 2004. — Vol. 53 (Suppl. 1). — P. 31–35.
63. Adiponectin — a key adipokine in the metabolic syndrome / J.P. Whitehead, A.A. Richards, I.J. Hickman // Diabetes Obes. Metab. — 2006. — Vol. 8, № 3. — P. 264–280.
64. Disruption adiponectin causes insulin resistance neo–intimal formation / N. Kubota, Y. Terauchi, T. Yamauchi [et al.] // J. Biol. Chem. — 2002. — Vol. 277. — P. 25863–25866.
65. Diet–induced insulin resistance in mice lacking adiponectin/ACRP30 / N. Maeda, I. Shimomura, K. Kishida [et al.] // Nat. Med. — 2002. — Vol. 8. — P. 731–737.
66. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity / T.P. Combs, U.B. Pajvani, A.H. Berg [et al.] // Endocrinology. — 2004. — Vol. 145. — P. 367–383.
67. Adiponectin stimulates glucose utilization and fatty–acid oxidation by activating AMP–activated protein kinase / T. Yamauchi, J. Kamon, Y. Minokoshi [et al.] // Nature Med. — 2002. — Vol. 8, № 11. — P. 1288–1295.
68. Adiponectin induces the anti–inflammatory cytokines IL–10 and IL–1RA in human leukocytes / A.M. Wolf, D. Wolf, H. Rumpold [et al.] // Biochem. Biophys. Res. Commun. — 2004. — Vol. 323. — P. 630–635.
69. Selective suppression of endothelial cell apoptosis by the high molecular weight form of adiponectin / H.N. Kobayashi, S. Ouchi, K. Kihara [et al.] // Circ. Res. — 2004. — Vol. 94, № 4. — P. 27–31.
70. Retraction / A. Fukuhara, M. Matsuda, M. Nishizawa [et al.] // Science. — 2007. — Vol. 318, № 5850. — P. 565.
71. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin / A. Fukuhara, M. Matsuda, M. Nishizawa [et al.] // Science. — 2005. — Vol. 307, № 5708. — P. 426–430.
72. Cloning and characterization of the cDNA encoding a novel human pre–B–cell colony–enhancing factor / B. Samal, Y. Sun, G. Stearns [et al.] // Mol. Cell. Biol. — 2007. — Vol. 14. — P. 1431–1437.
73. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties / A. R. Moschen, A. Kaser, B. Enrich [et al.] // J. Immunol. — 2007. — Vol. 178. — P. 1748–1758.
74. Plasma visfatin concentrations and fat depot–specific mRNA expression in humans / J. Berndt, N. Kloting, S. Kralisch [et al.] // Diabetes. — 2005. — Vol. 54. — P. 2911–2916.
75. Plasma visfatin levels in patients with newly diagnosed and untreated type 2 diabetes mellitus and impaired glucose tolerance / T. Dogru, A. Sonmez, I. Tasci [et al.] // Diabetes Res. Clin. Pract. — 2005. — Vol. 76. — P. 24–29.
76. Tilg H. Cytokines in alcoholic and non–alcoholic steatohepatitis / H. Tilg, A.M. Diehl // N. Engl. J. Med. — 2000. — Vol. 343. — Р. 1467–1476.
77. Михальчишин Г.П. Рівень чинника некрозу пухлин альфа і його кореляційні взаємозв’язки у хворих на цукровий діабет типу 2 із неалкогольною жировою хворобою печінки. / Г.П. Михальчишин, П.М. Боднар, Н.М. Кобиляк // Клінічна ендокринологія та ендокринна хірургія. — 2014. — № 1(46). — С. 33–40.
78. Tartaglia L.A. Two TNF receptors / L.A. Tartaglia, D.V. Goeddel // Immunol. Today. — 1992. — Vol. 13. — Р. 151–153.
79. Hotamisligil G.S. Adipose expression of tumor necrosis factor–alpha: direct role in obesity–linked insulin resistance / G.S. Hota–misligil, N.S. Shargill, B.M. Spiegelman // Science. — 1993. — Vol. 259. — Р. 87–91.
80. Protection from obesity–induced insulin resistance in mice lacking TNF–alpha function / K.T. Uysal, S.M. Wiesbrock, M.W. Marino [et al.] // Nature. — 1997. — Vol. 389. — Р. 610–614.
81. A central role for JNK in obesity and insulin resistance / J. Hirosumi, G. Tuncman, L. Chang [et al.] // Nature. — 2002. — Vol. 21, № 420. — P. 333–336.
82. Reversal of obesity– and diet–induced insulin resistance with salicylates or targeted disruption of Ikk–в / M. Yuan, N. Konstantopoulos, J. Lee [et al.] // Science — 2001. — Vol. 293. — P. 1673–1677.
83. Shoelson S.E. Inflammation and the IKK– /I–kB/NF–B axis in obesity– and diet–induced insulin resistance / S.E. Shoelson, J. Lee, M. Yuan // Int. J. Obes. Relat. Metab. Disord. — 2003. — Vol. 27. — P. 49–52.
84. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kB kinase complex / Z. Gao, D. Hwang, F. Bataille [et al.] // J. Biol. Chem. — 2002. — Vol. 277. — P. 48115–48121.
85. Local and systemic insulin resistance resulting from hepatic activation of IKK– and NF–kB / D. Cai, M. Yuan, D.F. Frantz [et al.] // Nat. Med. — 2005. — Vol. 11. — P. 183–190.
86. Mechanism by which high–dose aspirin improves glucose metabolism in type 2 diabetes / R.S. Hundal, K.F. Petersen, A.B. Mayerson [et al.] // J. Clin. Invest. — 2002. — Vol. 109. — P. 1321–1326.
87. Dinarello CA. Interleukin–1 in the pathogenesis and treatment of inflammatory diseases / C.A. Dinarello // Blood. — 2011. — Vol. 117. — P. 3720–3732.
88. Adipose and liver expression of IL–1 family members in morbid obesity and effects of weight loss / A.R. Moschen, C. Molnar, B. Enrich [et al.] // Mol. Med. — 2011. — Vol. 17, № 7–8. — P. 840–845.
89. Михальчишин Г.П. Рівень ІЛ–1в та його кореляційні взаємозв’язки у хворих на цукровий діабет типу 2 із неалкогольною жировою хворобою печінки. / Г.П. Михальчишин, П.М. Боднар, Н.М. Кобиляк // Ендокринологія. — 2013. — № 4. — С. 21–28.
90. Interleukin–1beta may mediate insulin resistance in liver–derived cells in response to adipocyte inflammation / O. Nov, A. Kohl, E.C. Lewis et al. // Endocrinology. — 2010. — Vol. 151. — P. 4247–4256.
91. Interleukin–1 receptor antagonist is upregulated during diet–induced obesity and regulates insulin sensitivity in rodents / E. Somm, P. Cettour–Rose, C. Asensio [et al.] // Diabetologia. — 2006. — Vol. 49. — P. 387–393.
92. XOMA 052, an anti–IL–1{beta} monoclonal antibody, improves glucose control and {beta}–cell function in the diet–induced obesity mouse model / A.M. Owyang, K. Maedler, L. Gross [et al.] // Endocrinology. — 2010. — Vol. 151. — P. 2515–2527.
93. Interleukin–1–receptor antagonist in type 2 diabetes mellitus / C.M. Larsen, M. Faulenbach, A. Vaag [et al.] // N. Engl. J. Med. — 2007. — Vol. 356. — P. 1517–1526.
94. A cysteine–rich adipose tissue–specific secretory factor inhibits adipocyte differentiation / K.H. Kim, K. Lee, Y.S. Moon, H.S. Sul // J. Biol. Chem. — 2001. — Vol. 276. — P. 11252–11256.
95. The hormone resistin links obesity to diabetes / C.M. Steppan, S.T. Bailey, S. Bhat [et al.] // Nature. — 2001. — Vol. 409. — P. 307–312.
96. Banerjee R.R. Resistin: molecular history and prognosis / R.R. Banerjee, M.A. Lazar // J. Mol. Med. — 2003. — Vol. 81. — P. 218–226.
97. Abnormal glucose homeostasis due to chronic hyperresistinemia / S.M. Rangwala, A.S. Rich, B. Rhoades [et al.] // Diabetes. — 2004. — Vol. 53. — 1937–1941.
98. Adipose–derived resistin and gut–derived resistin–like molecule–selectively impair insulin action on glucose production / M.W. Rajala, S. Obici, P.E. Scherer, L. Rossetti // J. Clin. Invest. — 2005. — Vol. 111. — P. 225–230.
99. Adenovirus–mediated chronic ‘hyper–resistinemia’ leads to in vivo insulin resistance in normal rats / H. Satoh, M.T. Nguyen, P.D. Miles [et al.] // J. Clin. Invest. — 2004. — Vol. 114. — P. 224–231.
100. Loss of resistin improves glucose homeostasis in leptin deficiency / Y. Qi, Z. Nie, Y.S. Lee [et al] // Diabetes. — 2006. — Vol. 55. — P. 3083–3090.
101. SOCS–3 inhibits insulin signaling and is up–regulated in response to tumor necrosis factor– in the adipose tissue of obese mice / B. Emanuelli, P. Peraldi, J. Filloux [et al.] // J. Biol. Chem. — 2001. — Vol. 276. — P. 47944–47949.
102. Nagaev J. Insulin resistence and type 2 diabetes are not related to resistin expression in human fat cells or skelet muscle / J. Nagaev, U. Smith // Biochem Biophys Re Commun. — 2001. — Vol. 285. — P. 561–564.
103. Increased resistin blood levels are not associated with insulin resistance in patients with renal disease / J.T. Kielstein, B. Becker, S. Graf [et al.] // Am. J. Kidney Dis. — 2003. — Vol. 42. — P. 62–66.
104. The interleukin–6 (–174) G/C promoter polymorphism is associated with type–2 diabetes mellitus in Native Americans and Caucasians / B. Vozarova, J.M. Fernandez–Real, W.C. Knowler [et al.] // Hum. Genet. — 2003. — V. 112. — P. 409–413.
105. 5–amino–imidazole carboxamide riboside acutely potentiates glucose–stimulated insulin secretion from mouse pancreatic islets by KATP channeldependent and independent pathways / C. Z. Wang, Y. Wang, A. Di [et al.] // Biochem. Biophys. Res. Commun. — 2005. — V. 330. — P. 1073–1079.
106. Interleukin–6 acts as insulin sensitizer on glycogen synthesis in human skeletal muscle cells by phosphorylation of Ser473 of Akt / C. Weigert, A.M. Hennige, K. Brodbeck [et al.] // Am. J. Physiol. Endocrinol. Metab. — 2005. — V. 289. — P. 251–257.
107. Pickup J. C. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes / J.C. Pickup // Diabetes Care. — 2004. — V. 27. — P. 813–823.
108. Circulating interleukin–6 in relation to adiposity, insulin action, and insulin secretion / B. Vozarova, C. Weyer, K. Hanson [et al.] // Obes. Res. — 2001. — V. 9. — P. 414–417.
109. Adipose tissue IL–6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro / J.P. Bastard, M. Maachi, J.T. Van Nhieu [et al.] // J. Clin. Endocrinol. Metab. — 2002. — V. 87. — P. 2084–2089.
110. Interleukin–6 and tumor necrosis factor — alpha are not increased in petients with Type 2 diabetes: evidence that plasma interleukin–6 is related to fat mass and not insulin responsiveness / A.L. Carrey, C.R. Bruce, M. Sacchetti [et al.] // Diabetologia. — 2004. — V. 47. — P. 1029–1037.
111. Rotter V. Interleukin–6 (IL–6) induces insulin resistance in 3T3–L1 adipocytes and is, like IL–8 and tumor necrosis factor–alpha, overexpressed in human fat cells from insulin–resistant subjects / V. Rotter, I. Nagaev, U. Smith // J. Biol. Chem. — 2003. — V. 278. — P. 45777–45784.
112. Adiponectin gene expression and secretion is inhibited by interleukin–6 in 3T3–L1 adipocytes / M. Fasshauer, S. Kralisch, M. Klier [et al.] // Biochem. Biophys. Res. Commun. — 2003. — V. 301. — P. 1045–1050.
113. Chronic exposure to interleukin–6 causes hepatic insulin resistance in mice / P.J. Klover, T.A. Zimmers, L.G. Koniaris, R.A. Mooney // Diabetes. — 2003. — V. 52. — P. 2784–2789.
114. Suppressor of cytokine signalling 3 expression and insulin resistance in skeletal muscle of obese and type 2 diabetic patients / J. Rieusset, K. Bouzakri, E. Chevillotte [et al.] // Diabetes. — 2004. — V. 53. — P. 2232–2241.
115. Interleukin–6–deficient mice develop mature–onset obesity / V. Wallenius, K. Wallenius, B. Ahren [et al.] // Nat. Med. — 2002. — V. 8. — P. 75–79.
116. Differential effects of interleukin–6 and–10 on skeletal muscle and liver insulin action in vivo / H.J. Kim, T. Higashimori, S.Y. Park [et al.] // Diabetes. — 2004. — V. 53. — P. 1060–1067.
117. Mooney R.A. Counterpoint: interleukin–6 does not have a beneficial role in insulin sensitivity and glucose homeostasis / R.A. Mooney // J. Appl. Physiol. — 2007. — V. 102. — P. 816–818.
118. Effect of endotoxin–induced monokines on glukose metabolism in the muscle cell line L6 / M.D. Lee, A. Zentella, W. Vine [et al.] // Proc. Natl. Alad. Sci. USA. — 1987. — V. 84. — P. 2590–2594.
119. Двойственная роль интерлейкина–6 в развитии инсулинорезистентности / В. Шварц // Патологическая физиология и экспериментальная терапия. — 2010. — № 1. — С. 40–47.