Статья опубликована на с. 130-139
Согласно современным представлениям, сахарный диабет 1-го типа (СД‑1) является хроническим аутоиммунным заболеванием, возникающим в результате комплексного взаимодействия генетической предрасположенности и факторов внешней среды, при котором происходит постепенное, селективное органоспецифическое снижение числа и функции инсулинпродуцирующих бета-клеток в поджелудочной железе [13]. Существует огромное количество работ и обзоров, в которых подчеркивается, что центральная роль в механизме развития СД‑1 принадлежит Т-клеточной системе адаптивного иммунитета [6, 13, 14, 17]. Однако роль клеточных элементов естественного иммунитета, особенно нейтрофилов, в патогенезе СД‑1 освещена в значительно меньшей степени. Вместе с тем в последние годы появился ряд публикаций, указывающих на существенное значение нейтрофилов в механизмах развития аутоиммунных процессов в организме человека, в частности в механизме развития СД‑1 [4, 5, 14, 30].
Нейтрофильные гранулоциты, получившие свое название от окраски их цитоплазматических гранул в нейтральный цвет по Гимза-Романовскому, иногда называют еще полиморфноядерными лейкоцитами, так как они характеризуются многодолевым сегментированным ядром. Зрелый нейтрофил здорового человека обычно содержит 4–5 сегментов. Наличие меньшего числа, особенно двух сегментов, характерно для аномалии Пельгера. Название «полиморфноядерные лейкоциты», по нашему мнению, не совсем точное, так как аналогичное многодолевое ядро имеют и другие типы гранулоцитов — эозинофилы и базофилы, содержащие в своей цитоплазме иные виды гранул, которые окрашиваются в красный или фиолетовый цвет.
Нейтрофилы представляют собой самую большую группу циркулирующих лейкоцитов у взрослого человека (т.е. 60–70 %, или 2/3 количества), что указывает на особую их роль в первой линии врожденной иммунной защиты организма от бактериальных и грибковых инфекций, а также стрессовых воздействий [3].
Нейтрофил очень подвижен. Он первый среди других видов лейкоцитов появляется в месте воспаления. Вместе с тем зрелый нейтрофил в циркулирующей крови является «дремлющей» конечной клеткой, так как не способен к делению и после выполнения своей героической функции по недопущению патогенов в организм сразу же погибает, больше не возвращаясь в циркуляцию [15, 19].
Характерной цитологической особенностью нейтрофила является наличие в его цитоплазме трех видов гранул. При светооптическом исследовании в окрашенных мазках крови различают: 1) первичные, или азурофильные, гранулы, самые крупные по размеру, окрашивающиеся азуром в синий цвет, которые появляются на стадии промиелоцита; 2) вторичные, или специфические, гранулы, менее крупные по размеру, при использовании краски азур/эозина окрашиваются в розово-фиолетовый (нейтральный) цвет, появляются на стадии миелоцита; 3) самые маленькие — третичные, или студенистые (gelatinase), гранулы обнаруживаются впервые только на стадии палочкоядерных форм [19, 20].
Нейтрофилы — это высокодифференцированные клетки, происходящие из общих с моноцитами клеток-предшественников (КОЕ-ГМ). В костном мозге цитологически различают шесть последовательных стадий созревания нейтрофилов: миелобласты, промиелоциты, миелоциты, метамиелоциты (юные), палочкоядерные и сегментоядерные нейтрофилы (рис. 1).
Процесс созревания нейтрофилов в костном мозге, который длится около двух недель, регулируется интерлейкином-3 и транскрипционным фактором С/ЕВР, экспрессированным преимущественно на незрелых миелоидных клетках [21]. После выхода из костного мозга зрелый нейтрофил циркулирует в крови около 12 часов, после чего мигрирует в ткани. Митотическая стадия созревания нейтрофила (миелобласт, промиелоцит и миелоцит) длится 7,5 дня, постмитотическая стадия (метамиелоцит, палочкоядерный и сегментоядерный нейтрофил) — 6,5 дня. Зрелый нейтрофил, поступивший в кровь, находится там всего 1–2 суток.
Основной функцией нейтрофила считается фагоцитоз. Процесс распознавания и обезвреживания нейтрофилами микробов, грибков и чужеродных клеток сложен и включает в себя хемотаксис, фагоцитоз и уничтожение бактерий, в нем принимают участие многие биологически активные соединения (лейкотриены, селектины, интегрины, опсонины, цитокины, хемокины и др.).
Вкратце этот процесс можно представить следующим образом. Инфицирование или воспаление приводит к стимуляции цитокинов ФНО-α, ИЛ-1β и ИЛ-17. Затем наступает экспрессия Р- и Е-селектинов и членов семейства инкретинов ICAM и VCAM на внутренней поверхности сосуда, находящегося в месте инфекции. Эндотелиальные клетки продуцируют хемокины ИЛ-8 и МIР-2, что приводит к стимуляции нейтрофилов и их хемотаксису. С помощью хемотаксиса осуществляется способность нейтрофилов обнаруживать микроорганизмы и целенаправленно двигаться по направлению к ним в очаг воспаления. Для этого у нейтрофила имеются специфические рецепторы для компонента С5а системы комплемента и протеаз, выделяемых при повреждении тканей или непосредственно бактериальным возбудителем, а также различных цитокинов и хемокинов. К очагу воспаления или повреждения тканей нейтрофилы продвигаются по градиенту хемотаксиса. Для этого они вращаются и катятся по эндотелиальной поверхности сосудов, замедляя свое движение относительно скорости всего потока крови в месте воспаления, где происходит их маргинация и прилипание к эндотелию активированного сосуда. В этом месте нейтрофил выпускает псевдоподий в направлении воспалительных хемотоксинов и проникает через сосудистую стенку. При этом в псевдоподий сначала поступает цитоплазма, а затем и ядро. Одновременно происходит опсонирование внедренного в организм патогена. Захват и поглощение опсонированных микроорганизмов осуществляются с помощью цитоплазматических пузырьков, которые превращаются в фагосомы. В дальнейшем эти фагосомы сливаются с первичными и вторичными нейтрофильными гранулами и происходит выход содержимого гранул (бактерицидных ферментов: лизоцима, миелопероксидазы, кислой и щелочной фосфатазы, эластазы, лактоферрина и др.) в фагосому, где наступает киллинг бактерии. Для переваривания и уничтожения патогенов нейтрофил генерирует высокотоксичный супероксид кислорода (О2) и перекись водорода (Н2О2) при активном участии в этом процессе NADPH-оксидазы. В результате происходит респираторный взрыв, при котором образуется более чем 50-кратное увеличение потребления кислорода по сравнению с нормой. Кроме выработки токсически активных форм кислорода бактерицидно губительным действием на патогены обладает и кислая среда фагосом, лизоцим, а также перечисленные выше бактерицидные белки дефензин и перфорин, которые приводят к расширению пор в поверхностных мембранах бактерии, что обусловливает более легкое проникновение в них бактерицидных веществ. Активация фагоцитоза усиливается под действием ФНО-α и ИФН-γ [3, 19].
Нейтрофилы, которые мигрируют в ткани, обладают более высокой фагоцитарной активностью, чем нейтрофилы крови. Тканевые нейтрофилы стимулируют транскрипционную программу, приводя к генерации ряда хемокинов, которые вызывают рекрутацию в очаг воспаления и повреждения тканей других видов воспалительных клеток — макрофагов и Т-клеток, которые составляют с нейтрофилами функциональные ассоциации.
В течение последних 10 лет была обнаружена и другая важная антимикробная функция нейтрофилов, а именно то, что эти клетки, кроме фагоцитоза, обладают также способностью ловить микроорганизмы с помощью экстрацеллюлярной ловушки (капкана), обозначаемой NETs (Neutrophil Extracellular Trap), что происходит на расстоянии, отдаленном от внутренней жизнедеятельности нейтрофила. NETs представляет собой сеть, сотканную из деконденсированных нитей ДНК, гистонов, протеинов цитоплазматических гранул и цитозоля на поверхности нейтрофила. При этом происходит разрушение ядра и растворение хроматина, которые совместно с протеинами гранул и цитозолем вытесняются на поверхность нейтрофила. Спектрофотометрически в этой субстанции определяется 24 вида протеинов (бактерицидные ДНК, связанные с белками, катепсин G, лактоферрин, миелопероксидаза и др.). Благодаря данной функции осуществляется как защитное, так и токсическое действие. Свойство нейтрофила убивать клетку с помощью ловушки NETs получило название нетоза в отличие от апоптоза и некроза [19, 30, 42]. Было также обнаружено, что особенно выраженной способностью к нетозу обладают нейтрофилы слизистой рта. Интересно отметить, что благодаря этой функции нейтрофилов осуществляется бактерицидный гомеостаз ротовой полости. Именно снижение NET-функции нейтрофилов слюны способствует развитию различных воспалительных заболеваний слизистой ротовой полости [33].
Однако роль нейтрофилов в организме человека не ограничивается только фагоцитозом и нетозом патогенов, она более полифункциональна. Нейтрофилы одновременно являются также секреторными клетками [46], продуцирующими в циркуляцию большое количество различных видов биологически высокоактивных соединений. С помощью секретируемых соединений нейтрофилы участвуют во многих реакциях адаптивного и естественного иммунитета [19, 20, 47].
Совсем недавно были описаны еще два пептида (NGAL и ADAM9), секретируемые нейтрофилами, которые обладают регуляторным действием на функцию капилляров [29]. Показано, что NGAL (Neutrophil Gelatinase-Association Lipocalin) оказывает индуцирующее влияние на дистальные тубулярные сегменты нефрона почек и может даже служить биологическим маркером острого почечного повреждения [30, 39].
ADAM9 (дизинтегрин и домен металлопротеиназы 9) способствует проницаемости альвеолярно-капиллярного барьера легких и резко изменяется при их остром воспалении [38].
Хотя роль цитокинов в механизме антимикробного и антивоспалительного действия нейтрофилов в организме человека довольно велика и имеются данные о наличии у нейтрофилов рецепторов ко многим цитокинам [5, 6, 14], тем не менее информация о продукции и секреции различных видов цитокинов и хемокинов нейтрофилами чрезвычайно скудна. Так, имеются отдельные сообщения о том, что на различных этапах хемотаксиса, при прохождении через стенку капилляров и при фагоцитозе патогенов в тканях нейтрофил выделяет цитокины ИЛ-1β, ИЛ-17, ИЛ-23, ФНО-α и хемокины ИЛ-8 и MIP-1 [19]. В культуре тканей нейтрофилов, выделенных из крови человека, особенно после их инкубации с липосахаридами, нейтрофилы секретируют ИЛ-10, ИЛ-12 и хемокин ИЛ-8. Причем степень секреции их значительно изменяется при развитии СД-1 и воспалении [24].
Значительную роль в выяснении функции нейтрофилов сыграло также исследование их субмикроскопического строения с использованием трансмиссионной и сканирующей электронной микроскопии высокого разрешения, а также ультраструктурной цитохимии, в результате чего была получена неоценимая информация о состоянии их ядра, цитоплазматических и мембранных структур [2, 11, 16, 18, 20, 40].
Особенно ценные сведения о функции нейтрофилов в последние годы были обнаружены при использовании методов электронной микроскопии совместно с биохимическими исследованиями различных их органоидов, особенно гранул, выделенных дифференциальным центрифугированием в градиенте плотности перколла [19, 20, 29].
В проведенных нами [6, 7] трансмиссионных электронно-микроскопических исследованиях экваториальных срезов сегментоядерных нейтрофилов ПК у здоровых детей и взрослых обоих полов этот вид лейкоцитов не имел существенных ультраструктурных отличий и выглядел как округлые или овальные клетки со значительным числом выростов эктоплазмы и микроворсинок на плазматической мембране (рис. 2). Доли ядра сегментированного зрелого нейтрофила чаще всего представлены как отдельные образования, поскольку соединяющие их перемычки не всегда попадают в плоскость среза.
Характерной особенностью нейтрофилов, как уже упоминалось, являются цитоплазматические гранулы, среди которых различают три различных вида: первичные, или азурофильные, вторичные, или специфические, и третичные, или секреторные, везикулы [19].
На субмикроскопическом и ультрацитохимическом уровнях первичные (азурофильные) гранулы выглядят более крупными по размеру и наиболее электронно-плотными. Они образуются на внутренней стороне аппарата Гольджи на стадии промиелоцита. Эти гранулы содержат миелопероксидазу, кислые гидролазы, эластазу и катепсин G, а также бактерицидные белки (дефенсины, сергоцидины и катемицидины), расширяющие поры стенок бактерии и, таким образом, повышающие действие бактерицидных протеинов. Вопрос о том, являются ли первичные гранулы истинными лизосомами, дискутируется, так как они не содержат характерных для лизосом других тканей лизосомально-ассоциированных протеинов (LAMP) [11, 19, 20].
Вторичные пероксидазоотрицательные гранулы (более мелкие и менее электронно-плотные) образуются на выпуклой стороне аппарата Гольджи на стадии миелоцита. Они содержат лактоферрин, лизоцим, нейтрофильный желатиназо-ассоциированный липокалин — NGAL (neutrophilgelatinase-associated lipocalin) — протеины, связанные с витамином В12, и коллагеназу [29, 31, 39]. Совсем недавно было установлено, что главным компонентом вторичных гранул является антимикробный пропептид hCAP‑18 (proLL‑37), который легко определяется в плазме крови человека, что дает возможность клинически определить дефицит этих гранул и правильно диагностировать вид нейтрофилопении у больных [47].
По мере созревания и дифференцировки количество азурофильных гранул в нейтрофилах уменьшается таким образом, что в зрелом нейтрофиле их содержится 1/3, в отличие от специфических гранул, количество которых по мере созревания увеличивается [12, 16, 20].
Третичные гранулы появляются на стадии палочкоядерных нейтрофилов. Третичные гранулы секретируют дизинтегрин и домен металлопротеиназу (ADAM9), хотя de novo его не продуцируют, а только накапливают в желатиназу гранул. Недавно была показана большая роль ADAM9 в регуляции альвеолярно-капиллярного барьера [38]. Зрелые нейтрофилы крови человека содержат все три вида гранул [19, 20].
В сегментоядерном нейтрофиле аппарат Гольджи небольшой, количество митохондрий невелико, эндоплазматический ретикулум скуден и количество свободных полирибосом уменьшено. Содержание глюкозы и гликогена (в виде β-гранул или гликосом) повышено [3, 7, 40]. На экваториальных срезах виден также клеточный центр и вариабельное число пузырьков (вакуолей) возле клеточной мембраны (рис. 2), которые, как уже указывалось, при захвате микробов и фагоцитозе сливаются с нейтрофильными гранулами, превращаясь в фаголизосомы, где происходит переваривание и уничтожение микробов.
Из всего вышеизложенного следует, что нейтрофилы являются лейкоцитами, которые путем фагоцитоза, нетоза и секреции бактерицидных веществ осуществляют первую линию защиты организма от вредоносных агентов, т.е. до того времени, когда в борьбу вступает адаптивная иммунная система. Однако при нарушении иммунной толерантности нейтрофилы способны оказывать и повреждающее действие на клетки собственного организма, в частности β-клетки [42] и клетки нефрона почек [39].
Роль нейтрофилов в патогенезе СД‑1 до недавнего времени практически целенаправленно не изучалась, так как основное внимание в этом вопросе уделялось исследованию адаптивного иммунитета (особенно различным субпопуляциям Т-лимфоцитов), лежащего в основе аутоиммунных процессов, характерных для этого заболевания.
Все же следует отметить, что исследования общего содержания нейтрофилов и их фагоцитарной активности в крови больных СД проводятся уже многие годы, но при этом были получены неоднозначные результаты. Это объясняется тем, что в более раннее время еще не существовало классификации СД, т.е. деления его на 1-й и 2-й тип, и исследования проводились на больных сахарным диабетом без учета его типа, возраста пациентов, гендерного различия, длительности заболевания и характера инсулинотерапии. Причем качество инсулина в то время было еще далеко до совершенства. Только в начале нынешнего столетия появилось несколько работ, достоверно показавших, что для больных ювенильным СД‑1 характерна небольшая нейтрофилопения. Так, в 2002 году нами впервые было показано [4], что детям с впервые выявленным СД‑1 (без специфических осложнений) присуще умеренное снижение абсолютного числа нейтрофилов в ПК, которое удерживается в течение более трех лет после установления диагноза и пребывания больных на хорошо контролируемой инсулинотерапии.
В дальнейших наших исследованиях [6] было обнаружено, что у здоровых детей с отягощенной наследственностью, но положительных не менее чем к двум островковым аутоантителам (ОАА) — IA‑2A и GADA, также наблюдается более низкое содержание абсолютного числа нейтрофилов в ПК по сравнению с ОАА-отрицательными и здоровыми детьми контрольных групп, что дает основания утверждать, что нейтрофилопения у «здоровых», но ОАА-положительных детей имеется задолго до возникновения СД‑1.
В 2013 году большая группа авторов [41] из различных клинических центров, принимающих участие в выполнении программы NHS, используя метод подсчета нейтрофилов с помощью гематологических анализаторов, подтвердила наши данные. Они показали, что у детей с недавно диагностированным СД‑1 имеется статистически достоверный (p < 0,0001) более низкий уровень нейтрофилов в ПК, чем у контрольной группы. Полученные данные подтверждались также обнаружением нейтрофилопении и у заболевших СД‑1 детей спустя год после установления диагноза. В работе A. Valle et al. (2013) приводятся также данные о том, что нейтрофилопения характерна не только для детей с уже диагностированным СД‑1, но и в доклинический бессимптомный период заболевания, т.е. когда дети еще здоровы, но с положительным тестом не менее чем к двум из четырех используемых ОАА (GADA, ICA, IA‑2A и ZnT8). В группе детей, отрицательных ко всем ОАА, абсолютное количество нейтрофилов в ПК было близким к норме.
В редакционной статье [37] авторитетного журнала Diabetes, в котором опубликована эта статья, утверждается, что полученные данные являются значительным открытием, «сюрпризом», наводящим на мысль, что в патогенезе ювенильного сахарного аутоиммунного диабета важное значение имеет не только адаптивный иммунитет, но и клеточные элементы врожденного иммунитета.
Причины и механизм снижения числа нейтрофилов в циркуляции у больных СД‑1 только выясняются. Полагают, что нейтрофилопения является следствием ухода нейтрофилов из ПК в поджелудочную железу в связи с развитием в ней воспалительного процесса [14, 30]. В пользу этого говорит обнаружение выраженной инфильтрации нейтрофилами островков Лангерганса (ОЛ) и экзокринной части поджелудочной железы человека, особенно в начальный период развития СД‑1 [22, 42]. Описана также выраженная инвазия в экзокринную часть поджелудочной железы больных СД‑1 клеток, экспрессирующих CD 11-антиген, который является маркером клеточных элементов естественного иммунитета: нейтрофилов, моноцитов и ЕК-клеток [36].
В то же время в исследованиях, проведенных на мышах, было показано [43], что при СД происходит блокада созревания незрелых миелоидных клеток Gr‑1+CD 11b+, включающих нейтрофилы в костном мозге (КМ), что обусловлено репрессией ключевого миелоидного транскрипционного фактора СЕВРА. Причем введение Г-СКФ приводит к восстановлению нормального нейтрофилопоэза у диабетических мышей.
Кроме того, большая группа финских генетиков [28], исследовавших с помощью транскрипционного анализа роль множества генов и молекулярных путей в патогенезе СД‑1, в доклинической (у ОАА-положительных лиц) и ранней клинической стадии развития этого заболевания, обнаружила диабетические изменения в транскрипции почти 100 генов. Особые нарушения были выявлены в модуле нейтрофила, заключающиеся в изменении регуляции липокалина‑2 и матрикса металлопротеазы в специфических гранулах, которые были активированы с помощью нейтрофил-ассоциированной транскрипции как у здоровых, но ОАА-положительных детей, так и после развития у них СД‑1.
В связи с убедительными доказательствами, что при СД‑1 имеется достоверная нейтрофилопения, естественно, возникает вопрос: сопровождается ли количественное уменьшение числа нейтрофилов их качественными изменениями?
Вместе с тем при исследовании функции нейтрофилов ПК у больных СД‑1 с помощью определения их фагоцитарной активности были получены не совсем однозначные результаты. Одна группа авторов [8, 27] наблюдала снижение фагоцитоза, в то время как другая [23, 44, 45] не обнаружила существенного изменения способности нейтрофилов захватывать микробы у пациентов с СД‑1 по сравнению со здоровыми людьми. Все же подавляющее большинство исследователей приходят к единому мнению, что у больных СД‑1 имеется выраженное снижение бактерицидной активности нейтрофилов, т.е. подавление внутриклеточного киллинга с помощью генерации высокотоксичных супероксида кислорода и перекиси водорода (респираторного взрыва), а также снижение активности бактерицидных соединений, находящихся в гранулах [19, 23, 26, 32, 34, 44, 45].
Как известно, СД‑1 способствует развитию тяжелых воспалительных осложнений и возникновению туберкулеза, грибковых и кожных инфекций. В механизме возникновения этих осложнений существенное значение, как полагают, имеет снижение фагоцитарной активности моноцитов/макрофагов и особенно нейтрофилов [10, 32, 35].
Как уже упоминалось ранее, нейтрофилы, наряду с фагоцитарной активностью, способны также захватывать и уничтожать патогены и инородные клетки с помощью специальной ловушки NET, расположенной на их поверхности, т.е. путем нетоза. Полагают, что гибель β-клетки при СД‑1 осуществляется нейтрофилами преимущественно путем нетоза. Так, недавно было показано [30, 42], что при нарушениях иммунной толерантности нейтрофилы благодаря NET могут принимать участие в механизме деструкции β-клеток выделенными на поверхности токсическими соединениями (эластазами и протеазами).
Хорошим подтверждением того, что нейтрофилы участвуют в механизмах патогенеза СД‑1, являются также недавно опубликованные данные [25] о том, что при испытании ряда лечебных препаратов, используемых при иммуноинтервенции СД‑1, введение колониестимулирующего фактора (G-CSF) у больных СД‑1 приводит к частичному сохранению функции β-клеток (снижение уровня HbA1c и С-пептида). Как известно, введение препаратов G-CSF вызывает резкое повышение числа нейтрофилов у человека [1, 5]. Это наводит на мысль, что благоприятное действие G-CSF при СД‑1 обусловлено устранением нейтрофилопении в результате стимуляции предшественников нейтрофилов в КМ и повышением их поступления в циркуляцию.
Как известно, одним из способов выяснения роли нейтрофилов является использование морфофункциональных методов их исследования, особенно электронной микроскопии и ультраструктурной цитохимии. Однако информация о функции нейтрофилов в крови у больных СД‑1, полученная с использованием метода электронной микроскопии, чрезвычайно ограниченна.
Впервые в литературе [4, 6] нами проведено изучение субмикроскопической организации нейтрофилов ПК при СД‑1 с помощью электронной микроскопии и ультраструктурной цитохимии на четырех группах детей: 1) практически здоровых без генетически отягощенной наследственности по СД‑1 (контроль); 2) здоровых, но с отягощенной наследственностью, отрицательных к ОАА; 3) здоровых с отягощенной наследственностью и положительных к ОАА; 4) больных СД‑1 (впервые выявленных и спустя 1–3 года после установления диагноза).
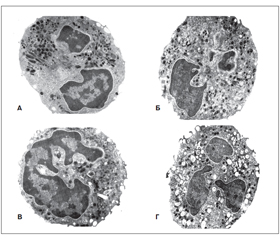
При исследовании ультратонких экваториальных срезов сегментоядерных нейтрофилов ПК под электронным микроскопом у ОАА-отрицательных детей с отягощенной наследственностью этот вид нейтрофилов по своей ультраструктуре был подобен таковым у здоровых лиц (рис. 3А, Б). Нейтрофилы ПК у ОАА-позитивных детей с отягощенной наследственностью по СД‑1 (рис. 3В) по своей ультраструктуре несколько отличались от таковых у детей контрольной группы и здоровых лиц с отягощенной наследственностью, но ОАА-отрицательных. Это также были округлые или овальные клетки со значительным числом микроворсинок на плазматической мембране. Ядро состояло из нескольких фрагментов, которые имели небольшие выросты различной конфигурации. Более выраженные изменения ультраструктуры нейтрофилов ПК по сравнению с нормой и ОАА-отрицательными детьми наблюдались со стороны цитоплазматических первичных пероксидазоположительных гранул, в которых иногда отмечались изменения деструктивного характера и резкое снижение активности пероксидазы. Это было особенно выражено у детей, у которых в последующем возник СД‑1, что наводит на мысль о том, что уже в этот доклинический период развития заболевания происходит выраженная дисфункция нейтрофилов.
/136.jpg)
У детей с только что возникшим СД‑1 до назначения им заместительной инсулинотерапии наблюдались более значительные изменения ультраструктуры нейтрофилов крови, чем у ОАА-положительных еще здоровых детей. Обнаруживалось большее число нейтрофилов с нарушенной структурой гранул в виде их набухания и деструкции, увеличенным размером митохондрий, фокальным расслоением ядерной мембраны. В ядрах наблюдались многочисленные выросты в виде шпилек и гантелей [4, 48].
Наиболее значительные изменения субмикроскопического строения нейтрофилов ПК были выявлены у детей с более длительным течением СД‑1, особенно более трех лет (рис. 3Г) после его диагностирования. В этой стадии течения СД‑1 в крови больных появлялось значительное количество нейтрофилов с характерным гладким контуром плазматических мембран. Ядро имело более извилистый контур, в результате чего в цитоплазме на срезах обнаруживались фрагменты ядра различных конфигураций в виде отдельных образований. Нередко отмечалось очаговое расширение ядерной мембраны с ее раздвоением и накоплением в этих местах рибосом, хотя общее их количество в клетке было сниженным. В цитоплазме многих клеток было видно набухание первичных и вторичных гранул, а также имелись гранулы с частичной их деструкцией и дегрануляцией. При ультрацитохимическом определении в первичных гранулах почти не определялась активность пероксидазы (рис. 4А, Б).
/137.jpg)
Иногда встречались участки цитоплазмы, лишенные органелл и заполненные скоплением остатков канальцев ГЭР, т.е. тельца Döhle (рис. 4В) [40], иногда обнаруживающиеся при сильной стрессовой реакции [9]. У значительной части детей, болеющих 3 и более 5 лет, некоторые нейтрофилы были сильно дегранулированными, так, что выглядели как дырчатые (рис. 3Г).
Полученные нами данные о значительном субмикроскопическом изменении нейтрофилов, особенно деструкции гранул, которые содержат многочисленные бактерицидные соединения, хорошо объясняют механизм описанного выше снижения киллерной активности этого вида лейкоцитов у больных СД‑1 и, следовательно, повышенной чувствительности пациентов к различным воспалительным осложнениям (диабетические нефропатия, ретинопатия, стопа и др., а также склонность к развитию других заболеваний) [35]. Особенно значительные изменения ультраструктуры и функции нейтрофилов при длительно текущем СД‑1 могут также служить одним из объяснений наблюдаемых при этом заболевании сосудистых осложнений, так как нейтрофилы одновременно с противомикробной защитной функцией принимают участие, согласно современным представлениям [9], также в регуляции эндотелия мелких сосудов и капилляров.
Особенно злободневным в настоящее время является вопрос об участии нейтрофилов в сложных механизмах, приводящих к снижению функции или к гибели инсулинпродуцирующих клеток поджелудочной железы у человека. В последних публикациях [17, 30] показано, что нейтрофилы у больных СД‑1 при нарушении иммунологической толерантности могут участвовать совместно с адаптивным иммунитетом в разрушении клеток собственного организма, в том числе β-клеток, подобно Т-лимфоцитам. Появились также веские доказательства прямой взаимосвязи нейтрофилов и адаптивной иммунной системы с β-клетками поджелудочной железы в механизме их апоптоза и нетоза при СД‑1. Так, показано [14, 37], что аутореактивные Т-клетки и островковые β-клеточные антигены на поверхности β-клеток находятся в функциональной ассоциации с клетками естественного иммунитета (нейтрофилами, макрофагами, дендритными клетками). Подвергшиеся триггерам β-клетки продуцируют хемокины СХСL10, CCL2 и цитокин ИФН-γ, что вызывает аттракцию как адаптивных, так и естественных иммунных клеток к панкреатическим ОЛ, индуцируя их воспаление. Предполагается, что нейтрофилы могут либо прямо действовать на β-клетки, либо активировать дендритные клетки, особенно посредством аутоантител, которые, в свою очередь, стимулируют адаптивную иммунную систему. Эти данные согласуются с работами, в которых показано, что у больных СД‑1 повышена инфильтрация нейтрофилами островков Лангерганса и экзокринной части поджелудочной железы, особенно на начальной стадии возникновения заболевания [14, 22].
Таким образом, анализируя приведенную выше литературу и собственные данные, можно прийти к заключению о значительной роли нейтрофилов при СД‑1 и его осложнениях у человека, что обусловлено рядом факторов:
1. Снижением у больных СД‑1 антимикробной защиты организма в результате уменьшения содержания нейтрофилов в ПК и ослабления их функции (фагоцитоз, нетоз и секреция антибактерицидных соединений нейтрофильными гранулами путем экзоцитоза в циркуляцию). Ухудшение защиты организма от патогенов является также одной из главных причин воспалительных осложнений, характерных для СД‑1, и повышения склонности к развитию других болезней. Подтверждением этого являются данные о том, что восстановление содержания нейтрофилов в ПК у больных СД‑1 с нейтрофилопенией приводит к улучшению концентрации HbA1c в ПК и клинического состояния пациента.
2. Нарушением секреции нейтрофилами иммунорегуляторных соединений, контролирующих функцию сердечно-сосудистой системы, особенно мелких сосудов и капилляров, что приводит к развитию ангиопатий у больных СД‑1.
С другой стороны, установлено, что нейтрофилы способны также оказывать деструктивное действие на β-клетки, что обусловлено нарушением иммунологической толерантности при СД‑1. В результате этого нейтрофилы, подобно Т-лимфоцитам, могут участвовать в аутоиммунных реакциях, приводящих к разрушению собственных β-клеток, что согласуется с обнаружением значительной инфильтрации нейтрофилами экзокринной части поджелудочной железы и ОЛ у человека. Показано также, что аутореактивные Т-клетки, макрофаги и нейтрофилы образуют в поджелудочной железе взаимосвязанные функциональные конгломераты.
Новая информация о значительной роли нейтрофилов в патогенезе СД‑1 указывает на то, что в механизме развития этого заболевания наряду с адаптивным иммунитетом принимают участие и клеточные элементы естественного иммунитета. Дальнейшее изучение этого вопроса будет способствовать разработке новых, более эффективных способов профилактики и лечения этого тяжелого заболевания и его осложнений.
Список литературы
1. Бутенко А.К., Афанасьева В.В., Глуховская И.Ю., Климнюк Г.И., Балицкая О.В., Зак К.П. Ультраструктурная характеристика лейкоцитов крови при лечении рчГ-КСФ (филграстимом) химиотерапевтической миелосупрессии у детей с неходжкинскими лимфомами // Эксперим. онкология. — 1997. — Т. 19, № 2. — С. 147-152.
2. Бутенко З.А., Глузман Д.Ф., Зак К.П. и др. Цитохимия и электронная микроскопия клеток крови и кроветворных органов / Под ред. В.Г. Пинчука; Ин-т пробл. онкологии, Киев. НИИ эндокринологии и обмена веществ. — К.: Наук. думка, 1974. — 247 с.
3. Дейл М.М. Нейтрофильные лейкоциты // Руководство по иммунофармакологии / Под ред. М.М. Дейла, Дж.К. Формена. — М.: Медицина, 1998. — 331 с.
4. Зак К.П., Малиновская Т.Н., Тронько Н.Д. Иммунитет у детей, больных сахарным диабетом. — К.: Книга плюс, 2002. — 111 с.
5. Зак К.П., Попова В.В. Иммунная интервенция в терапии сахарного диабета (аналитический обзор) // Диабет. Ожирение. Метаболический синдром. — 2015. — № 6 (IV). — С. 31-44.
6. Зак К.П., Тронько Н.Д., Попова В.В., Бутенко А.К. Сахарный диабет. Иммунитет. Цитокины. — К.: Книга-плюс. — 2015. — 485 с.
7. Комиссаренко С.В., Зак К.П. Радиация и иммунитет человека. — К.: Наукова думка, 1994. — 111 с.
8. Окороков А.И., Селиванов В.Н., Немцов В.M. и др. Функциональное состояние лейкоцитов у больных сахарным диабетом // Терапевтический архив. — 1982. — Т. 54, № 10. — С. 27-30.
9. Шиффман Ф. Дж. Патофизиология крови. — М.: Бином, 2009. — 446 с.
10. Abrass C.K. Fc-receptor-mediated phagocytosis: abnormalities associated with diabetes mellitus // Clin. Immunol. Immunopathol. — 1991. — Vol. 58, № 1. — P. 1-17.
11. Ackerman G.A. The human neutrophilicmyelocyte. A correlated phase and electron microscopic study // Z. Zellforsch. Mikrosk. Anat. — 1971. — Vol. 121, № 2. — P. 153-170.
12. Ackerman G.A. The human neutrophilicpromyelocyte. A correlated phase and electron microscopic study // Z. Zellforsch. Mikrosk. Anat. — 1971. — Vol. 118, № 4. — P. 467-481.
13. Atkinson M.A., Eisenbarth G.S., Michels A.W. Type 1 diabetes // Lancet. — 2014. — Vol. 38, № 9911. — P. 69-82.
14. Atkinson M.A., von Herrath M., Powers A.C., Clare-Salzler M.Current concepts on the pathogenesis of type 1 diabetes — considerations for attempts to prevent and reverse the disease // Diabetes Care. — 2015. — Vol. 38, № 6. — P. 979-988.
15. Baggiolini M., Dewald B. The neutrophil // Int. Archs Allergy and Appl. Immunology. — 1985. — Vol. 76, Suppl. 1. — P. 13-20.
16. Bainton D.F., Ullyot J.L., Farquhar M.G. The developmentof neutrophilicpolymorphonuclear leukocytes in human bone marrow // J. Exp. Med. — 1971. — Vol. 134, № 4. — P. 907-934.
17. Battaglia M., Atkinson M.A. The streetlight effect in type 1 diabetes // Diabetes. — 2015. — Vol. 64, № 4. — P. 1081-1090.
18. Bessis M. Living blood cells and their ultrastructure. — New York; Heidelberg; Berlin: Springer-Verlag, 1973. — 239 p.
19. Borregaard N. Neutrophils, from marrow to microbes // Immunity. — 2010. — Vol. 33, № 5. — P. 657-670.
20. Cieutat A.M., Lobel P., August J.T. et al. Azurophilic granules of human neutrophilic leukocytes are deficient in lysosome-associated membrane proteins but retain the mannose 6-phosphate recognition marker // Blood. — 1998. — Vol. 91, № 3. — P. 1044-1058.
21. Combart A.F. The end of line for neutrophils // Blood. — 2015. — Vol. 125, № 11. — P. 1688-1690.
22. Diana J., Simoni Y., Furio L. et al. Crosstalk between neutrophils, B‑1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes // Nat. Med. — 2013. — Vol. 19, № 1. — P. 65-73.
23. Dziatkowiak H., Kowalska M., Denys A. Phagocytic and bactericidal activity of granulocytes in diabetic children // Diabetes. — 1982. — Vol. 31, № 12. — P. 1041-1043.
24. Glowacka E., Banasik M., Lewkowicz P., Tchorzewski H. The effect of LPS on neutrophils from patients with high risk of type 1 diabetes mellitus in relation to IL‑8, IL‑10 and IL‑12 production and apoptosis in vitro // Scand. J. Immunol. — 2002. — Vol. 55, № 2. — P. 210-217.
25. Haller M.J., Gitelman S.E., Gottlieb P.A. et al. Anti-thymocyte globulin/G-CSF treatment preserves β cell function in patients with established type 1 diabetes // J. Clin. Invest. — 2015. — Vol. 125, № 1. — P. 448-455.
26. Herskowitz I.C., Matsutani A., Permutt M.A. Reversibility of respiratory burst dysfunction in the human diabetic neutrophil is dependent on tight control of the diabetic state // Diabetes. — 1991. — Vol. 40, Suppl. 1. — P. 475A.
27. Hisashi N., Yuichi H., Moriyasu T. et al. Фагоцитарная и бактерицидная активность полиморфноядерных лейкоцитов у больных инсулинзависимым сахарным диабетом (ИЗСД). Связь между функцией полиморфноядерных нейтрофилов и контролем гликемии // J. Jap. Diabet. Soc. — 1991. — Vol. 34, № 1. — P. 7-13.
28. Kallionpää H., Elo L.L., Laajala E. et al. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility // Diabetes. — 2014. — Vol. 63, № 7. — P. 2402-2414.
29. Kjeldsen L., Bainton D.F., Sengeløv H., Borregaard N. Identification of neutrophil gelatinase-associated lipocalin as a novel matrix protein of specific granules in human neutrophils // Blood. — 1994. — Vol. 83, № 3. — P. 799-807.
30. Leslie R.D., Bradford C. Autoimmune diabetes: Caught in a NET // Diabetes. — 2014. — Vol. 63, № 12. — P. 4018-4020.
31. http://www.hindawi.com/journals/ije/2015/765364/Lucisano S., Arena A., Stassi G. et al. Role of paricalcitol in modulating the immune response in patients with renal disease role of paricalcitol in modulating the immune response in patients with renal disease // International J. Endocrinology. — 2015. Article ID 765364, 8 pages.
32. Miller M. Phagocytic cell functions in diabetes mellitus // Immunol. Clin. and Exp. Diabetes. — New York; London, 1984. — P. 369-383.
33. Mohanty T., Sjögren J., Kahn F. et al. A novel mechanism for NETosis provides antimicrobial defense at the oral mucosa // Blood. — 2015. — Vol. 126, № 18. — P. 2128-2137.
34. Nielson C.P., Hindson D.A. Inhibition of polymorphonuclear leukocyte respiratory burst by elevated glucose concentrations in vitro // Diabetes. — 1989. — Vol. 38, № 8. — P. 1031-1035.
35. Pisarczyk-Wiza D., Zozulinska D., Majchrzak A. et al. The influence of postprandial state on the polymorphonuclear neutrophils function in type 1 diabetic patients // Diabetologia. — 2002. — Vol. 45, Suppl. 2 (A‑36), 101.
36. Rodriguez-Calvo T., Ekwall O., Amirian N. et al. Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes // Diabetes. — 2014. — Vol. 63, № 11. — P. 3880-3890.
37. Röep B.O. β-cells, autoimmunity, and the innate immune system: un menage a trois // Diabetes. — 2013. — Vol. 62. — P. 1821-1822.
38. Roychaudhuri R., Hergrueter A.H., Polverino F. et al. ADAM9 is a novel product of polymorphonuclear neutrophils: regulation of expression and contributions to extracellular matrix protein degradation during acute lung injury // J. Immunol. — 2014. — Vol. 193, № 5. — P. 2469-2482.
39. Schilder L., Nurmohamed S.A., ter Wee P.M. et al. The plasma level and biomarker value of neutrophil gelatinase-associated lipocalin in critically ill patients with acute kidney injury are not affected by continuous venovenous hemofiltration and anticoagulation applied // Clinical Care. — 2014, 18 April R 78.
40. Tanaka Y., Goodman J.R. Electron microscopy of human blood cells. — New York; London: Harper at Row. Publ., 1972. — 432 p.
41. Valle A., Giamporcaro G.M., Scavini M. et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes // Diabetes. — 2013. — Vol. 62, № 6. — P. 2072-2077.
42. Wang Y., Xiao Y., Zhong L. et al. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with β-Cell autoimmunity in patients with type 1 diabetes // Diabetes. — 2014. — Vol. 63, № 12. — P. 4239-4248.
43. Wicks K., Torbica T., Umehara T. et al. Diabetes inhibits Gr‑1+ myeloid cell maturation via Cebpa deregulation // Diabetes. — 2015. — Vol. 64, № 12. — P. 4184-4197.
44. Wierusz-Wysocka B., Wysocki H., Wykretowicz A. et al. Phagocytosis, bactericidal capacity, and superoxide anion (O2–) production by polymorphonuclear neutrophils from patients with diabetes mellitus // Folia Haematol. Int. Mag. Klin. Morphol. Blutforsch. — 1985. — Vol. 112, № 5. — P. 658-668.
45. Wilson R.M., Reeves W.G. Neutrophil phagocytosis and killing in insulin-dependent diabetes // Clin. Exp. Immunol. — 1985. — Vol. 63, № 2. — P. 478-483.
46. Wright D.G. The neutrophil as a secretory organ of host defense. — In: Advances in host defense mechanisms: Phogocytic cells / J.I. Gallin, A.S. Fauci (eds.). — New York: Raven Press, 1982. — Vol. 1. — P. 75-110.
47. Ye Y., Carlsson G., Karlsson-Sjöberg J.M. et al. The antimicrobial propeptidehCAP‑18 plasma levels in neutropenia of various aetiologies: a prospective study // Sci. Rep. — 2015 Jun 29. — 5. — 11685. doi: 10.1038/srep11685.
48. Zak K.P., Afanasyeva V.V., Yefimov A.S., Tkach S.N. Ultrastructure of neutrophils of non treated patients with newly diagnosed IDDM and effects of human insulin therapy: 15 International Diabetes Federation Congress (November 6-11, 1994). — Kobe, Japan. — 07A5PP0362.
49. Zak K.P., Filatova R.S., Afanasyeva V.V. Cytochemical and ultracytochemical studies of peroxidase activity in rabbit blood granulocytes under hydrocortisone effect // Folia Haematol. Int. Mag. Klin. Morphol. Blutforsch. — 1983. — Vol. 110, № 4. — P. 490-502.

/131.jpg)
/133.jpg)