Статья опубликована на с. 140-151
В практической деятельности врача-эндокринолога нередко встречаются заболевания щитовидной железы (ЩЖ), которые характеризуются комбинацией клинических проявлений, свойственных нескольким заболеваниям этого органа. При этом оба заболевания в одном органе существуют одновременно, однако в клинической картине, как правило, доминирует одна болезнь [14]. В других случаях клинические проявления отдельных заболеваний у одного и того же больного калейдоскопически сменяют друг друга на протяжении длительного периода наблюдения, т.е. одно заболевание постепенно трансформируется в другое [24, 27].
Если у больного имеется достаточно клинических и серологических признаков, чтобы диагностировать конкретное аутоиммунное заболевание ЩЖ, но вдобавок к этому еще присутствуют симптомы другого заболевания, речь идет о так называемом синдроме перекреста (overlap-синдром). Несмотря на то что проявления обеих болезней могут наблюдаться одновременно, обычно симптоматика одного заболевания превалирует над симптомами другой болезни. Аутоиммунный синдром перекреста заболеваний ЩЖ — практически неизученная, хотя нередко встречающаяся патология. Оverlap-синдром в литературе определяют как две болезни одного органа, причем оба заболевания могут носить аутоиммунный характер или одно из них может быть вирусным или иного генеза [25].
Аутоиммунные заболевания ЩЖ составляют львиную долю в структуре заболеваний этого органа, и именно они чаще сочетаются в виде синдрома перекреста. Аутоиммунные заболевания эндокринных органов могут быть как органоспецифическими болезнями, так и проявлениями системных заболеваний, т.е. органонеспецифическими заболеваниями с плавным переходом. При органоспецифических аутоиммунных заболеваниях эндокринных органов чаще всего поражается ЩЖ, островковый аппарат поджелудочной железы и надпочечники. Как правило, аутоиммунный процесс ведет к частичной или полной деструкции эндокринного органа с последующим возникновением секреторной недостаточности. Отмечается четкая корреляция начала заболевания (даже при отсутствии симптоматики) с лимфоцитарной инфильтрацией. В последующем происходит разрушение клеточных структур с фиброзированием и в конечном итоге органной недостаточностью. На этой стадии активного иммунологического процесса довольно часто определяются антитела против определенных антигенов тканей-мишеней [1, 4, 6].
Патогенетические особенности развития аутоиммунных заболеваний ЩЖ позволяет выделить несколько типов заболеваний этого органа. По ведущему механизму развития аутоиммунную патологию ЩЖ можно разделить на две группы: заболевания, обусловленные аутоиммунной деструкцией гормонопродуцирующих клеток; синдромы нарушения функции, когда аутоиммунный процесс сопровождается стимуляцией или, наоборот, блокадой эндокринной функции.
Аутоиммунные заболевания ЩЖ представляют собой группу аутоиммунных тиреопатий, патогенетическую основу которых составляет генетически обусловленный дефект иммунной толерантности в отношении антигенов ЩЖ [1, 37]. С одной стороны, антигенная структура ЩЖ включает более 10 специфических антигенов, в то время как антигенный полиморфизм является фактором аутосенсибилизации и аутоагрессии. К настоящему времени клонированы 3 основных органоспецифических антигена: пероксидаза ЩЖ (тиреопероксидаза), тиреоглобулин и рецептор к тиреотропному гормону (ТТГ), к которым образуются антитела. Важную роль в патогенезе аутоиммунных заболеваний ЩЖ могут играть и другие тиреоидные антигены — тиреоидный транспортер йодида, второй коллоидный антиген и антиген, ассоциированный с аутоиммунным поражением ЩЖ.
Причинами развития аутоиммунных процессов при заболеваниях ЩЖ могут быть нарушения физиологической изоляции антигенов ЩЖ (по отношению к которым в организме нет иммунологической толерантности, а образующиеся при этом антитела обладают выраженной органной специфичностью); изменение антигенных свойств ЩЖ в результате повреждения (особенно часто в этом отношении обсуждается вопрос роли вирусов); прием препаратов йода, усиливающих дефект ЩЖ (если таковой имеется), связанный с органификацией йода; генетическая предрасположенность (изменения в лимфоидной системе). По мнению большинства авторов, генетическая предрасположенность играет главную роль в развитии аутоиммунных процессов [29]. Данные популяционных, генетических и иммуногенетических исследований позволяют считать, что основой аутоиммунных заболеваний ЩЖ в большей степени является биологическая детерминанта, чем социальная. То есть основным этиологическим фактором служит наследственность, в то время как средовые факторы играют второстепенную роль, способствуя реализации генотипа и модулируя аутоиммунный процесс [17]. С точки зрения формальной генетики аутоиммунные заболевания ЩЖ относятся к мультифакториальным заболеваниям. Характерными признаками мультифакториальной патологии являются генетическая гетерогенность и нечеткость нозологических определений, что на практике означает непредсказуемость типа наследуемости, особенностей клинического течения и исхода болезни. Нозологическая неопределенность, присущая аутоиммунным заболеваниям ЩЖ, затрудняет их распознавание и разработку методов лечения.
Современные иммуногенетические исследования подтвердили особую роль главного комплекса гистосовместимости (ГКГ) — антигенов ІІ класса в патогенезе аутоиммунных заболеваний ЩЖ. Антигены ГКГ ответственны не только за предрасположенность к развитию заболеваний, но и за сроки возникновения, характер течения и исход патологического процесса [21]. Получены данные о генетически обусловленной гетерогенности клинических вариантов диффузного токсического зоба (ДТЗ). Согласно результатам недавнего популяционного исследования, проведенного в Дании с участием 9 000 близнецов, от 64 до 90 % риска развития ДТЗ объясняется генетическим риском [17].
Ранние иммунологические и последующие генетические исследования показали устойчивую ассоциацию аллели DR 3 (DRB 1*0304) и аллели DQA1*0501 и ДТЗ. Относительный риск развития ДТЗ за счет ассоциации с ДR 3 составляет 2,0–3,5. Согласно анализу сцеплений область ГКГ определяет до 20 % семейной кластеризации заболевания [29]. Интерес представляет тот факт, что установлена взаимосвязь аутоиммунного тиреоидита (АИТ) с аллелями DR 3. Анализ генетического сцепления в 56 семьях с ДТЗ позволил обнаружить три локуса предрасположенности к данной патологии: их обозначили GD 1; GD 2; GD 3. Локусы сцепления и аллельной ассоциации ДТЗ на хромосоме 18q21 частично перекрываются с локусом предрасположенности к сахарному диабету 1-го типа. Взаимное перекрытие локусов предрасположенности и к ДТЗ, и к СД 1-го типа выявлены на хромосоме Хр11 [17].
Многообразие вариантов клинического течения аутоиммунных заболеваний ЩЖ связано с полиморфизмом генов. Каждый ген имеет множество вариаций. Полиморфизм генов подразделяют на структурный и функциональный. Структурный полиморфизм означает изменение последовательности на участках гена, чаще транскрипционных (экзонах), которое приводит к изменению аминокислотной последовательности в синтезируемом белке. Функциональный полиморфизм — это изменение последовательности чаще в некодирующихся частях гена (интронах), сигнальном пептиде, промоторе, тандемных повторах с изменяющимся числом копий (т.н. VNTR), результатом которого является изменение скорости синтеза белка. Полиморфизм, определение вариантов которого основано на названиях рестриктаз, использующихся для его типирования, получил название рестрикционного.
Исходя из вышесказанного, причиной аутоагрессии, а также факторами, определяющими локализацию аутоиммунного процесса, его конкретный механизм, особенности клинической манифестации, исход болезни и ее распространенность в популяции и относительный риск для родственников или носителей определенных генотипов, являются многочисленные условно-патогенные аллели ГКГ или других частей генома. Прежде всего это подразумевает полиморфизм генетической основы, которую весьма трудно уточнить. Генетические и иммуногенетические исследования позволяют сделать следующие выводы. С генетической и патогенетической позиции аутоиммунные заболевания ЩЖ не являются самостоятельными нозологическими формами. Это синдромы с многообразными причинами и механизмами развития, которые требуют дальнейшего изучения. Типирование ГКГ в качестве инструмента научных исследований и метода диагностики конкретных заболеваний не обладает достаточной чувствительностью и специфичностью. Этот метод позволяет достаточно успешно выявлять (идентифицировать) группы риска и прогнозировать клиническое течение заболевания. Общая иммунологическая основа аутоиммунных заболеваний ЩЖ затрудняет их нозологическую диагностику и тем самым демонстрирует отчетливую тенденцию к взаимотрансформации или последовательному или одновременному развитию у одного и того же больного. Отсюда трудность идентификации аутоиммунных заболеваний ЩЖ. Это является одной из причин того, что до сих пор нет единой общепризнанной классификации заболеваний ЩЖ. В 1997 г. R. Volpe предложил классификацию заболеваний ЩЖ.
Классификация заболеваний ЩЖ (Volpe R., 1997)
І. Аутоиммунные заболевания ЩЖ.
1. Аутоиммунный гипертиреоз (болезнь Грейвса).
2. Аутоиммунный тиреоидит (тиреоидит Хашимото, фиброзный вариант, лимфоцитарный тиреоидит детского и юношеского возраста, послеродовой «немой» тиреоидит; бессимптомный аутоиммунный тиреоидит).
ІІ. Неиммуногенные заболевания ЩЖ со вторичными иммунными реакциями: подострый тиреоидит Де Кервена, узловой зоб, папиллярный рак ЩЖ.
Еще одну классификацию аутоиммунных заболеваний ЩЖ предложили Davis и соавт. (1993). Эта классификация имеет прагматическую направленность в отличие от этиопатогенетической классификации R. Volpe (1997), и демонстрирует многовариантность течения ДТЗ и шаткость нозологического подхода при диагностике аутоиммунных заболеваний ЩЖ.
Классификация аутоиммунных заболеваний ЩЖ (Davis и соавт., 1993)
1. Аутоиммунный тиреоидит 1-го типа.
2. Аутоиммунный тиреоидит 2-го типа (транзиторный тиреоидит; с зобом (болезнь Хашимото); без зоба (атрофический тиреоидит).
3. Болезнь Грейвса (3-го типа): гипертиреоидная болезнь Грейвса; эутиреоидная болезнь Грейвса; гипотиреоидная болезнь Грейвса.
Весьма интересная, с практической направленностью классификация исключительно АИТ предложена Г.С. Зефировой (1999). В классификации выделяется АИТ как самостоятельная нозологическая форма и вариант течения в сочетании с другими заболеваниями (т.н. оverlap-синдром, хотя этот термин не используется).
Классификация аутоиммунного тиреоидита (Зефирова Г.С., 1999)
І. По функциональному состоянию (гипотиреоз, эутиреоз, тиреотоксикоз.
ІІ. По размеру щитовидной железы (гипертрофическая форма, атрофическая форма).
ІІІ. По нозологическому признаку: АИТ как самостоятельное заболевание; АИТ как сочетание с другой тиреоидной патологией (подострым тиреоидитом, узловым зобом, эндокринной офтальмопатией); АИТ как компонент аутоиммунного полигландулярного синдрома.
Между всеми вариантами тиреоидита, по-видимому, существуют какие-то генетические и/или патогенетические различия. Однако у них много общего, что позволяет объединять их под одним названием «АИТ». Большинство специалистов в области тиреоидологии считают, что обязательным признаком АИТ является лимфоцитарная инфильтрация ткани ЩЖ. Для обоснованного диагноза должна быть отчетливая очаговая или диффузная лимфоцитарная инфильтрация. В зависимости от характеристики интенсивности лимфоцитарной инфильтрации некоторые авторы выделяют следующие гистологические варианты АИТ [24]: классический лимфоцитарный тиреоидит (с обильной диффузной или очаговой инфильтрацией лимфоцитами, множеством своеобразных герминативных центров из клеток Ашкенази, макрофагов и фибробластов), лимфоматозный тиреоидит (с менее выраженной лимфоцитарной инфильтрацией преимущественно зрелыми лимфоцитами), атрофический (фиброзный) АИТ (характеризуется резким фиброзом трабекул, стромы при относительно небольшой лимфоцитарной инфильтрации).
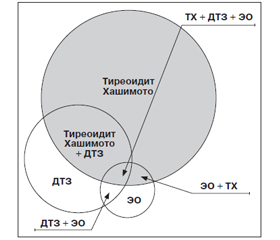
Многие иммунологические и гистологические аспекты ДТЗ сходны с таковыми при классическом АИТ, и эти заболевания нередко встречаются в одной и той же семье. Имеется ряд описаний монозиготных близнецов, один из которых страдает ДТЗ, а другой — АИТ. Более того, оба процесса могут развиваться в одной и той же железе [13, 37]. Клинические проявления в таких случаях зависят от преобладания того или иного из этих процессов. Лимфоидная инфильтрация ЩЖ так же, как и при АИТ, — одна из характерных особенностей морфологической картины нелеченого ДТЗ. Лимфоидная инфильтрация ЩЖ у больных ДТЗ может быть диффузной или очаговой, что значительно затрудняет возможность ее выявления путем пункционной биопсии или гистологического исследования. Когда лимфоидная инфильтрация достигает степени с формированием крупных лимфоидных фолликулов, то говорят о хашимотизации процесса ДТЗ, т.е. о ДТЗ с сопутствующим АИТ. Хашимотизация ДТЗ обусловливает спонтанное исчезновение тиреотоксикоза (спонтанное излечение от ДТЗ), причем у некоторых больных в дальнейшем даже развивается гипотиреоз. Последний регистрируется у 15 % больных ДТЗ без всякого лечения [21]. Это происходит за счет нарастания сопутствующего тиреоидита [18, 19]. Наличие выраженной лимфоидной инфильтрации ЩЖ у больных ДТЗ является, вероятно, благоприятным в отношении рецидива прогностическим признаком. Многие исследования не выявили лимфоидной инфильтрации ни у одного больного с рецидивом ДТЗ [30]. Кроме того, подчеркивалось, что больные ДТЗ, у которых пункция ЩЖ обнаруживала лимфоидную инфильтрацию, не склонны к рецидивированию после лечения тиреостатиками. Особенно часто при ДТЗ возникают т.н. фокальные тиреоидиты, которые, можно сказать, являются неотъемлемой частью этого заболевания. Они проявляются образованием в ЩЖ круглоклеточных инфильтратов и протекают так же, как и АИТ с образованием антител. Уровень содержания антител не достигает таких величин, как при тиреоидите Хашимото, так как клеточная инфильтрация незначительна. Поэтому фокальные тиреоидиты являются одной из разновидностей аутоиммунных тиреоидитов с незначительной клинической симптоматикой, которые, вероятно, являются причиной спонтанной ремиссии ДТЗ (процесс самоизлечения). Фокальные тиреоидиты нередко встречаются и при других заболеваниях ЩЖ — раке ЩЖ, ДТЗ и особенно у женщин после наступления климакса, которые клинически не проявляются, хотя лабораторно обнаруживают наличие антител [5]. Возникновение ДТЗ и сопутствующего АИТ в случае overlap-синдром можно представить следующим образом (рис. 1).
/143.jpg)
У человека клетки ЩЖ при ее аутоиммунных заболеваниях сами по себе нормальны и признаки их генетических нарушений отсутствуют.
В литературе нет убедительных данных об иммунологически значимых изменениях тиреоидных антигенов при аутоиммунных заболеваниях ЩЖ. Тиреоидная клетка является пассивным объектом иммунологической атаки, оставаясь внутренне нормальной.
T. Wilkin (1991) считает, что аутоиммунную агрессию следует рассматривать как физиологическую реакцию нормальной иммунной системы на аутоантигены, высвобождающиеся при воздействии какого-либо триггерного фактора, — вирусное воспаление или на какой-то другой антиген, экспрессируемый тканью-мишенью. По данным М.Э. Фисфален и Л. Дe Грут (2003), триггерными факторами активации Т-клеток при аутоиммунных заболеваниях ЩЖ могут быть следующие агенты (рис. 2).
При вирусных заболеваниях аутоиммунные заболевания развиваются лишь в очень редких случаях. Для того чтобы это произошло, необходимы нарушения в самой иммунной системе. Чрезмерная презентация антигена недостаточна для возникновения аутоиммунного заболевания ЩЖ. Они развиваются вследствие первичных нарушений иммунорегуляции. Дисфункция органа является результатом атаки недостаточно супрессированных антигенспецифических лимфоцитов, направленной против антигенов определенных клеток-мишеней.
Последовательность развития аутоиммунной болезни следующая. В результате нарушения иммунологического надзора, связанного с дефицитом Т-супрессоров, происходит размножение запрещенных клонов лимфоцитов, которые, с одной стороны, при значительной продукции ТСА приводят к развитию ДТЗ с более или менее выраженным развитием вначале фокального АИТ. С другой — преимущественное образование антител против антигенов ЩЖ способствует развитию АИТ. Приведенная схема довольно четко объясняет не только механизм одновременного наличия ДТЗ и АИТ, но и позволяет понять те наблюдения, когда один из монозиготных близнецов страдает ДТЗ, а другой заболевает АИТ. Это явление можно квалифицировать как факт иммуногенетической дивергенции, т.е. расщепление нозологических форм. Достоверно установлено, что ДТЗ и АИТ влияют друг на друга, однако до сих пор не установлено, насколько АИТ модифицирует течение ДТЗ. Следовательно, как явствует из вышеприведенной схемы, аутоиммунный синдром перекреста при заболеваниях ЩЖ представляет собой пример иммунологической конвергенции — сочетания (слияния) отдельных нозологических форм.
Таким образом, прогноз ДТЗ зависит от степени лимфоидной инфильтрации ЩЖ (что раньше называлось хашимотизацией процесса). Показателем степени этого процесса еще является титр антител к тиреоглобулину и микросомальной фракции (Yoshidа с соавт., 1978). Высокий титр антител всегда свидетельствует о наличии выраженной лимфоидной инфильтрации у больных ДТЗ, что подтверждает их прогностическое значение. Некоторые авторы полагают, что больных ДТЗ при наличии высокого титра антител следует не оперировать, а лечить консервативно, чтобы избежать весьма вероятного быстрого развития гипотиреоза. Клинический опыт свидетельствует, что такие больные не склонны к рецидивированию ДТЗ после отмены антитиреоидных препаратов.
Интерес представляет вопрос о влиянии субтотальной резекции ЩЖ на течение ДТЗ. Уже в 1960–70-е годы многочисленными исследованиями установлено, что субтотальная тиреоидэктомия не оказывает существенного влияния на аутоиммунный процесс. Этот вывод достаточно правомерен, основную причину появления соответствующих антител следует усматривать в первичном нарушении иммунного статуса, а не в появлении необычных для него антигенов. Причины заключаются в следующем. В течение долгого времени считалось, что для возникновения аутоиммунного процесса достаточно простого контакта белков ЩЖ с иммунокомпетентными клетками, в результате чего образуются органоспецифические аутоантитела, вызывающие повреждения ЩЖ. Однако, как показывают клинические наблюдения, кратковременное поступление в кровь тиреоидных антигенов при оперативных вмешательствах, кровоизлияниях в ЩЖ, подостром тиреоидите, некрозах обычно не приводит к развитию АИТ. Проблема оказалась более сложной, чем казалось на первый взгляд. Для АИТ характерно наличие высокого титра антител, однако патологические изменения в ЩЖ неизменно связаны с инфильтрацией лимфоцитами и плазматическими клетками. Следовательно, оперативное вмешательство само по себе не является фактором, способствующим возникновению стойкой аутоиммунной реакции. Послеоперационная динамика титров антител может быть связана с транзиторным изменением после операции количества антигена. Хирургическое лечение заболеваний ЩЖ не устраняет аутоиммунных нарушений и ведет к преходящему изменению количества антигена и титра антител.
То же касается иммунологических сдвигов при подостром тиреоидите Де Кервена. Однако большинство авторов считает эти нарушения вторичными и не имеющими большого значения в патогенезе. Сочетание ДТЗ и АИТ является закономерным явлением. Другой вопрос — степень выраженности того или иного процесса и, соответственно, превалирования клинической симптоматики одного из этих заболеваний. По-видимому, хашитоксикоз в его изначальном понимании (как лимфоидная инфильтрация ЩЖ) — один из закономерных возможных вариантов морфологической эволюции (исхода) ДТЗ.
Требует уточнения само понятие «хашитоксикоз», которое в последнее время изменилось. Изначально под термином «хашитоксикоз» подразумевали форму ДТЗ с выраженной лимфоидной инфильтрацией, затем — периоды тиреотоксикоза у больных АИТ, позже — сочетание у одного и того же больного морфологических признаков ДТЗ и АИТ (overlap-синдрома), а в последнее время — волнообразное течение АИТ со сменами периодов гипо- и тиреотоксикоза. В сущности, все состояния, которые обозначались раньше и сейчас термином «хашитоксикоз», полностью соответствуют понятию «аутоиммунный синдром перекреста» при заболеваниях ЩЖ. У некоторой части больных АИТ в дебюте возможно наличие тиреотоксикоза, который некоторые авторы связывают с процессом деструкции ткани ЩЖ вследствие аутоагрессии и поступлением в кровь большого количества ранее синтезированных гормонов [9, 11]. Однако при АИТ не бывает такого масштабного одномоментного разрушения ткани ЩЖ, которое способствовало бы, как при подостром тиреоидите, выбросу в кровь значительного количества гормонов в течение довольно длительного времени. Вероятно, возможной причиной преходящего тиреотоксикоза может быть наличие антител, стимулирующих продукцию тиреоидных гормонов. Однако в дальнейшем в связи с деструкцией ткани ЩЖ и, соответственно, количества функционирующей активной ткани ЩЖ значение этих антител уменьшается.
Как и другие аутоиммунные заболевания, ДТЗ и АИТ имеют волнообразное течение с фазами ремиссии и обострения. При ДТЗ в ЩЖ обнаруживается поликлональный Т-клеточный ответ, причем речь идет о гетерогенности как Т-клеток, так и аутоантигенов. До 80 % больных ДТЗ имеют антитела, которые реагируют с тиреоидной пероксидазой, в несколько меньшем проценте случаев встречаются антитела к тиреоглобулину. Сам тиреоцит может вносить прямой вклад в прогрессирование аутоиммунного процесса. Индивидуальные особенности регуляции этих процессов являются как минимум одной из детерминант различного клинического течения ДТЗ, в частности его тяжести, размера зоба и прогноза тиреостатической терапии. С учетом возможности попеременной продукции различных вариантов антител к рецептору ТТГ — блокирующих и стимулирующих, что у больного может сопровождаться сменой гипо- и гипертиреоза, становится понятным, что рецептор ТТГ и антитела к нему обладают исключительными и необычными свойствами, изучение которых приблизит нас к выяснению патогенеза ДТЗ [33, 36]. Имеются сообщения о том, что даже при длительно существующем гипотиреозе функция ЩЖ может спонтанно нормализоваться [24]. Подобное изменение функционального состояния ЩЖ можно объяснить появлением ранее отсутствующих тиреостимулирующих антител. Другая версия заключается в том, что изначально имеющиеся блокирующие антитела к рецептору ТТГ меняют свой характер (происходит спонтанная реверсия) — точку приложения на рецепторе ТТГ, становясь стимулирующими антителами. Кроме того, продукция антител может носить временный характер. В этом случае секреция гормонов ЩЖ у больных может возвращаться к норме. Больные с таким волнообразным течением заболевания являются наиболее сложной группой, т.к. требуют постоянного контроля и изменения тактики лечения.
Аутоиммунные заболевания щитовидной железы (ДТЗ и АИТ) и эндокринная офтальмопатия (ЭО) могут наблюдаться в отдельности или в любом сочетании, и их изучение представляет интерес с точки зрения overlap-синдрома. В зарубежной литературе ЭО носит название «офтальмопатия Грейвса». Известно, что ЭО может развиться до появления тиреотоксикоза, одновременно с ним, и во время лечения [2, 3]. Около 40 % больных ДТЗ не имеют проявлений ЭО. Описаны случаи ее появления при АИТ, а также у больных без признаков какого-либо заболевания ЩЖ, ни клинических, ни лабораторных, т.е. на фоне эутиреоза [7, 9]. В последнем случае она получила название эутиреоидной болезни Грейвса. В настоящее время имеются убедительные доказательства аутоиммунной природы ЭО [15, 28]. Наиболее важным, исходя из принципа сходства, является, очевидно, то, что природа ЭО и аутоиммунных заболеваний ЩЖ одинакова. Авторы ряда работ пытаются объяснить взаимосвязь между патологией ЩЖ и ЭО. D. Weightmann и соавт. (1989) показали, что мышцы глазных яблок имеют сродство к тиреоглобулину и к иммунным комплексам «тиреоглобулин-антитело». По их данным, существуют лимфатические связи между ЩЖ и ретробульбарной областью. На основании этого авторы постулировали, что транспорт по лимфатической системе тиреоидных антител, комплексов «антиген-антитело», иммунокомпетентных клеток к ретробульбарному пространству ведет к развитию ЭО. Другие работы подчеркивают сродство мышц глазного яблока к комплексу «тиреоглобулин-антитиреоглобулин». При ДТЗ и ЭО в иммунный процесс вовлекаются разные популяции лимфоцитов. Из этого следует, что ЭО представляет собой отдельное заболевание, часто сочетающееся с ДТЗ. В фазе развития иммунологических нарушений, несмотря на то что в основе развития ДТЗ и АИТ лежит органоспецифический ответ, наблюдается также поликлональная активация иммунной системы, сопровождающаяся образованием антител к клеткам и структурам, не имеющим прямого отношения к тиреоглобулину, тиреоидной пероксидазе, β-клеткам поджелудочной железы, клеткам гипофиза и ретробульбарным клеткам [21]. Это хорошо отражает схема патогенеза аутоиммунных заболеваний ЩЖ, предложенная L. De Groot и соавт. (табл. 1).
/146.jpg)
Сходные иммунологические нарушения наблюдаются и при других аутоиммунных эндокринопатиях (например, СД 1-го типа).
Гистологические исследования нарушений при ЭО показали наличие инфильтрации ретробульбарных мышц с герминативными центрами, хотя не обнаружено антител к тканям глаза [15]. В зависимости от того, какой тип изменений обнаруживается в острой фазе болезни, различают [2]: ЭО с поражением преимущественно глазодвигательных мышц; ЭО с поражением клетчатки; ЭО смешанного генеза.
Если патогенетические механизмы развития ДТЗ и ЭО являются аутоиммунными, что лежит в их основе? Прежде всего, наследственная предрасположенность. Роль наследственности подтверждается такими фактами, как агрегация ДТЗ у родственников, выраженная конкордантность у монозиготных близнецов, высокая частота других семейных органоспецифических аутоиммунных заболеваний. Несмотря на генетическую предрасположенность к ДТЗ, до сих пор неясно, почему фенотипические проявления не наблюдаются у всех восприимчивых особей. Наиболее высокая заболеваемость наблюдается в возрасте 30–45 лет.
Представляет интерес вопрос о ведущих причинах аутоиммунного конфликта при ЭО.
На протяжении десятилетий медицинская статистика накопила данные, свидетельствующие о том, что первая манифестация многих аутоиммунных заболеваний эндокринной системы, и в том числе ЩЖ, происходит после ряда вирусных, микробных и паразитарных инфекций. В ретробульбарной и тиреоидной ткани больных с офтальмопатией найдены IgM и IgA, реагирующие с антигенами Yersinia enterolitika [14]. Кроме того, эти суперантигены индуцируют и экспрессию молекул ГКГ ІІ класса на ретробульбарных фибробластах. Антигены к некоторым микробным антигенам, например Yersinia enterolitika, перекрестно реагируют с тиреоидными антигенами, что может свидетельствовать о связи ДТЗ с этим энтеропатогенным агентом. Эндогенные суперантигены кодируются ретровирусами, а экзогенные представлены разнообразными белками бактериального и вирусного происхождения.
Детальный анализ этих причинно-следственных отношений привел к проблеме т.н. молекулярной мимикрии возбудителя инфекций. Диверсия возбудителя осуществляется под камуфляжем структур, химически родственных антигенам тех или иных экзокринных желез. Это общебиологическое явление оказалось особенно характерным для патогенеза СД 1-го типа. То же может иметь место и при ДТЗ с ЭО. Общие антигенные детерминанты возбудителя и тиреоцитов, ретробульбарной клетчатки, глазодвигательных мышц после инфекционного эпизода оставляют пролонгированный аутоиммунный конфликт. Следовательно, манифестацию ЭО в сочетании с заболеванием ЩЖ можно связать с развитием аутоиммунного ответа на эпитопы микроорганизмов и перекрестных реакций со сходными эпитопами тканей ЩЖ, ретробульбарной клетчатки, глазодвигательных мышц (рис. 3). Эпитопы — часть антигена, взаимодействующая с антителами.
На рис. 4 представлены взаимоотношения между ДТЗ, АИТ и ЭО.
Возможен и иной механизм участия бактериальной и вирусной инфекции в возникновении ЭО [9, 15, 35]. Под действием триггерных механизмов (бактериальная и вирусная инфекции, токсины, курение, радиация) у генетически предрасположенных лиц на мембранах глазодвигательных мышц и других тканей орбиты происходит усиление экспрессии аутоантигенов. Имеющийся дефект Т-супрессоров способствует выживанию и размножению Т-хелперов, направленных против аутоантигенов ЩЖ и тканей орбиты. Дефект иммунологического надзора усугубляется при тиреотоксикозе. В ответ на появление аутоантигенов Т-лимфоциты и макрофаги инфильтрируют мышцы орбиты, высвобождая цитокины. Последние стимулируют пролиферацию фибробластов ретробульбарного интерстиция, выработку гликозаминогликанов и коллагена.
/147_2.jpg)
Практически одновременное развитие тиреотоксикоза и офтальмопатии у большинства больных может свидетельствовать в пользу единых иммунологических механизмов. Перекрестным реагированием тиреоидных антител с мышцами орбиты можно объяснить корреляцию между тяжестью тиреотоксикоза и поражения глаз. В настоящее время связь ЭО с патологией ЩЖ объясняется не столько значимостью комплексов, содержащих дериваты ТТГ и антитела к ТГ, сколько наличием цитотоксических антител, дающих перекрестную реакцию с поверхностным антигеном мышц глаза и клеток ЩЖ [7, 28]. Антитела были обнаружены у 60 % лиц, страдающих ДТЗ и ЭО, и у 80 % лиц с ЭО, протекающей на фоне АИТ [33]. При обследовании пациентов только с патологией ЩЖ без ЭО титры цитотоксических антител были низкими. Вероятно, существует поверхностный антиген, имеющий общий эпитоп с антигенами глазодвигательных мыщц. У лиц с генетической склонностью к заболеванию рецепторы ТТГ экспрессируются на жировых клетках, включая и те, что находятся в орбите. При наличии аутоиммунной реакции образовавшиеся антитела к рецептору тиреоцитов могут реагировать с ТТГ-рецепторами жировой ткани орбиты [35]. В результате развивается воспалительный процесс, который сопровождается поступлением в ретробульбарную область мононуклеарных клеток, в том числе аутореактивных В-лимфоцитов. Таким образом, патогенетические факторы ЭО множественные и имеют отношение ко всей группе аутоиммунных заболеваний ЩЖ — ДТЗ и АИТ. Каждое из заболеваний может проявляться не только самостоятельно, но и в паре или триадой, чем и объясняется различное состояние функции ЩЖ при ее аутоиммунных нарушениях.
Клиническая манифестация поражений ЩЖ и орбиты, т.е. фенотипические проявления, особенности течения, темпа прогрессирования, зависят от иммуногенетической структуры синдрома. Если преобладает продукция ТСА, то развивается гипертрофическая форма АИТ, и, напротив, преобладание антител, блокирующих ТТГ-рецепторы, приводит к возникновению атрофического варианта заболевания. В ряде случаев дебют АИТ может протекать с признаками гиперфункции ЩЖ. При этом особую актуальность приобретает вопрос дифференциальной диагностики между началом ДТЗ и АИТ или сочетания этих заболеваний, т.е. overlap-синдрома, или конверсии одного заболевания в другое. Эти трудности дифференциальной диагностики ДТЗ и АИТ на ранних стадиях могут сохраняться до тех пор, пока болезнь не завершится тем или иным исходом.
Заключение
АИТ и ДТЗ рассматриваются как самостоятельные нозологические единицы. Вместе с тем существуют варианты течения заболеваний, при которых одновременно присутствуют признаки как АИТ, так и ДТЗ, что в известной степени стирает границы между этими аутоиммунными поражениями ЩЖ. У этой категории больных наряду с клиническими и/или гистологическими особенностями, характерными для АИТ, выявляются признаки, свойственные ДТЗ. По патогенезу между этими заболеваниями больше сходства, чем различий. Логика рассуждений и фактические данные приводят к необходимости выделения т.н. overlap-синдрома (синдрома перекреста). Больные с синдромом перекреста имеют крайне пеструю клиническую, биохимическую и серологическую картину.
Развитие аутоиммунного синдрома перекреста при заболеваниях ЩЖ, вероятно, связано с общей иммунологической основой, одним из компонентов является главный комплекс гистосовместимости. Выделение overlap-синдрома важно для практического врача не только в плане правильной постановки и формулировки диагноза, но и для выработки наиболее рациональной терапевтической тактики.
Иллюстрацией overlap-синдрома при заболеваниях ЩЖ могут послужить два нижеприведенных наблюдения.
Клинический случай 1
Больная С.Н.,1973 г.р. Диагноз ДТЗ был выставлен в феврале 1999 г. Со стороны ЩЖ отмечалось диффузное увеличение (зоб ІІ ст.). До ноября 1999 г. получала адекватную тиреостатическую терапию тиамазолом (начальная доза 30 мг/сут). После отмены препарата клинически сохранялся эутиреоз. Через некоторое время при отсутствии контроля в середине 2005 года установлены проявления субклинического гипотиреоза и обнаружены аутоантитела к ТПО в титре 1: 500 ЕД. Поставлен диагноз АИТ. Назначено лечение LТ4 в дозе 50 мкг/сут. В дальнейшем в связи с беременностью больная находилась под тщательным контролем эндокринолога. Доза LТ4 увеличена до 100 мкг/сут. После рождения здорового сына в 2005 году ТТГ составлял 0,06 мМЕ/л и LТ4 отменили. В дальнейшем из-за развития гипотиреоза возобновлен прием LТ4 в дозе 50 мкг/сут. Данные обследования: состояние удовлетворительное. Рост 170 см. Вес 69 кг. АД 130/85 мм рт.ст. ЧСС 80 в 1'. Кожа обычной влажности. Глазные симптомы отсутствуют. ЩЖ не пальпируется. Рефлексы одинаковы с обеих сторон. Лабораторные данные: 09. 2006: ТТГ — 13 мМЕ/л, свТ4 — 12 пмоль/л, свТ3 — 3,0 пмоль/л, антитела к ТПО — 3600 ЕД/л, антитела к рецепторам ТТГ — 5,6 МЕ/л. Диагноз: ДТЗ (дебют с тиреотоксикоза). В процессе течения болезни развился латентный гипотиреоз в результате трансформации (перетекания) в АИТ. Течение и лечение: после повышения дозы LТ4 до 75 мг/сут достигнуто состояние эутиреоза. В дальнейшем в связи с развитием ремиссии LТ4 отменен. В 2008 г. при последнем контроле клинически состояние эутиреоза. Содержание ТТГ — 0,8 мМЕ/л. Обсуждение: три болезни у одной пациентки: ДТЗ — дебют с тиреотоксикоза, АИТ и эутиреоидная форма ДТЗ. Имеет ли место трансформация (переход) АИТ в ДТЗ? Тиреотоксическая форма ДТЗ может, как правило, через длительный промежуток времени — 10–15 лет — трансформироваться в гипотиреоз [9, 11, 12]. При этом патогенез этого явления может быть мультифакториальным. С одной стороны, описан деструктивный фокальный или диффузный тиреоидит (трансформация в классическую форму АИТ), с другой — доминирование блокирующих антител к рецептору ТТГ [12] (рис. 1). У пациентки речь идет об особом течении тиреотоксической формы ДТЗ после латентного периода — 6 лет — и его перехода в АИТ (антитела к ТПО — 3600 ЕД/л) с развитием латентного гипотиреоза. В дальнейшем у больной наблюдалась спонтанная ремиссия. Через 10 лет после начала заболевания отмечено развитие эутиреоидной формы ДТЗ, при этом уровень ТТГ находился в пределах нижней границы нормы. Примечательно то, что в начале развития ДТЗ не обнаруживались антитела к рТТГ и трудно было его дифференцировать от чистого АИТ с хашитоксикозом. Потребность в высоких дозах тиреостатиков в течение 9 месяцев и дальнейшее выявление антител к рТТГ свидетельствовали о ДТЗ. Недостаточно ясным является механизм развития гипотиреоидной формы ДТЗ во второй фазе заболевания. Резюмируя вышеизложенное, можно сказать, что у этой пациентки, вероятно, тиреотоксическая форма ДТЗ после латентного периода — 6 лет — трансформировалась в АИТ, а последний через 4 года опять перешел в эутиреоидную форму ДТЗ.
Клинический случай 2
Больная С., 59 лет. Жалобы и анамнез: в течение нескольких лет беспокоят сухость кожи, слабость, запоры. В последнее время состояние ухудшилось: появились зябкость, сонливость и прибавка массы тела (6 кг). Отмечает отсутствие потливости при физической нагрузке. Обратилась к участковому врачу, который направил на консультацию к эндокринологу. Объективно: рост 167 см, масса тела 68 кг. ЧСС 60 в 1'. АД 160/90 мм рт.ст. Кожа сухая, шелушится. Щитовидная железа не пальпируется. Сухожильные рефлексы снижены. Глазные симптомы отсутствуют. Лабораторно-инструментальные данные: ТТГ — 55 мМЕ/л, свТ4 — 4 нмоль/л, свТ3 — 8 нмоль/л, антитела к рТТГ — 10 ЕД/л, антитела к ТПО — 40 Ед/л. Диагноз: АИТ с исходом в гипотиреоз. Течение заболевания и лечение: LТ4 50 мкг/сут. Месяц спустя доза препарата увеличена до 75 мкг/сут. Через 2 месяца показатели функции ЩЖ: ТТГ — 30 мМЕ/л, свТ4 — 5,4 нмоль/л, свТ3 — 3,0 нмоль/л. В связи с сохранением состояния декомпенсации доза LТ4 увеличена до 100 мкг/сут. Через 1,5 месяца определение ТТГ и тиреоидных гормонов выявило латентный тиреотоксикоз: ТТГ — 0,08 мМЕ/л, свТ4 — 25 нмоль/л, свТ3 — 6,0 нмоль/л. Доза LТ4 уменьшена до 75 мкг/сут. Спустя два месяца сохранились проявления субклинического тиреотоксикоза (ТТГ — 0,15 мМЕ/л), в связи с чем проведено дальнейшее уменьшение дозы LТ4 до 50 мкг/сут, а затем — до 25 мкг/сут. В общей сложности через 8 месяцев после заместительной терапии достигнут эутиреоз. За это время масса тела уменьшилась на 5 кг. Обсуждение. Дифференциальная диагностика в данном случае должна проводиться между классической формой АИТ с исходом в гипотиреоз и т.н. гипотиреоидной формой ДТЗ. В основе патогенеза АИТ лежит преимущественно деструктивный аутоиммунный процесс. Признаком этого аутоиммунного процесса, т.е. повреждения паренхимы ЩЖ, является повышение титра антител к ТПО. Эти антитела выявляют в 95 % всех случаев АИТ и являются диагностически значимыми [10]. Широкому кругу врачей-специалистов мало или почти ничего не известно о существовании и роли нестимулирующих или блокирующих антител к рецептору ТТГ, которые были описаны в конце 1950-х годов. Они могут участвовать в развитии гипотиреоза при АИТ, и их выявляют в 10–20 % всех случаев болезни. Блокирующие антитела играют роль в развитии т.н. гипотиреоидной формы ДТЗ. У пациентов с этой формой болезни обнаруживают как стимулирующие, так и блокирующие антитела [9], исходя из гипотезы баланса. У данной пациентки имеется манифестный гипотиреоз, который ввиду высокого титра антител к рТТГ возник в связи с аутоиммунным процессом в ЩЖ. Титр антител к ТПО не был увеличен. Дифференциальную диагностику следует проводить между АИТ с исходом в гипотиреоз и т.н. гипотиреоидной формой ДТЗ. Поскольку у 5 % пациентов с АИТ антитела к ТПО не выявлены, то их отсутствие при лабораторных исследованиях не исключает абсолютно АИТ. Высокий титр антител к рТТГ, напротив, свидетельствует о ДТЗ. В данном случае имеется дисбаланс между стимулирующими антителами и блокирующими. Для верификации диагноза важно было бы определение блокирующих антител с помощью биологических методов, однако, к сожалению, таких методов в распоряжении врачей в настоящее время не имеется. Выявление блокирующих антител в общем для лечения не имеет значения, т.к., несмотря на это, в данном случае необходима заместительная терапия LT4.
Таким образом, вышеприведенные случаи из практики характеризуются комбинацией клинических проявлений, свойственных нескольким заболеваниям ЩЖ. При этом оба заболевания в одном органе существуют одновременно. Однако в клинической практике, как правило, доминирует одна болезнь (Фисфален М., Де Грут Л., 2003; Генделека Г.Ф., 2010). В первом случае клинические проявления одной болезни у одного и того же больного сменяют друг друга на протяжении длительного периода наблюдения, т.е. одно заболевание трансформируется в другое. Речь идет о т.н. синдроме перекреста (overlap-синдроме). Несмотря на то что проявления обеих болезней могут наблюдаться одновременно, обычно симптоматика одного заболевания превалирует над симптомами другой болезни. Аутоиммунный синдром перекреста ЩЖ — нередкая, хотя недостаточно изученная патология этого органа и terra incognita как для исследователей, так и для практикующих врачей.
Список литературы
1. Балаболкин М.И. Состояние и перспективы изучения проблемы физиологии и патологии щитовидной железы // Тер. архив. — 1997. — № 10. — C. 5-11.
2. Генделека Г.Ф. Аутоиммунный синдром перекреста (overlap-синдром) при заболеваниях щитовидной железы // Международный эндокринологический журнал. — 2010. — № 2. — C. 118-128.
3. Генделека Г.Ф. Атипичные формы течения аутоиммунных заболеваний щитовидной железы как проявление аутоиммунного синдрома перекреста (overlap-синдрома) // Международный эндокринологический журнал. — 2014. — № 3. — С. 103-107.
4. Дедов И.И., Трошина Е.А., Антонова С.С. и др. Аутоиммунные заболевания щитовидной железы: состояние проблемы // Пробл. эндокринол. — 2002. — № 2. — С. 7-12.
5. Зефирова Г.С. Заболевания щитовидной железы. — М.: Арт-Бизнес-Центр, 1999. — 215 с.
6. Лукьянчиков В.С., Калинин А.П., Лукьянчиков В.В. Современные представления об этиологии и патогенезе аутоиммунных эндокринопатий // Тер. архив. — 1995. — № 10. — С. 3-6.
7. Мазуров В.И., Святова Л.Е, Климко Н.Н. Современные представления о патогенезе, диагностике и лечении эндокринных офтальмопатий // Межд. медицин. обзоры. — 1993. — № 3. — С. 143-148.
8. Павлова Т.Л., Котова Г.А., Герасимова Г.А. Эндокринная офтальмопатия // Пробл. эндокринол. — 1998. — № 2. — С. 22-27.
9. Петунина Н.А. Клиника, диагностика и лечение аутоиммунного тиреоидита // Пробл. эндокринол. — 2002. — 36. — С. 16-21.
10. Родионова Т.И. Патогенез, диагностика и лечение эндокринной офтальмопатии // Пробл. эндокринол. — 1997. — № 6. — С. 46-56.
11. Смирнов В.В., Мавричева И.С. Тиреоидиты у детей // Лечащий врач. — 2006. — № 3. — С. 69-72.
12. Фадеев В.В. Болезнь Грейвса // Рус. мед. журнал. — 2005. — № 6. — С. 353-356.
13. Вольпе Р. Аутоиммунные заболевания щитовидной железы // Болезни щитовидной железы: Пер. с англ. — М., 2000. — С. 140-172.
14. Фисфален М. — Э., Де Грут Л. Болезнь Грейвса и аутоиммунный тиреоидит // Молекулярная эндокринология: Пер. с англ. / Под ред. Б.Д. Вайнтрауба. — M.: Mедицина, 2003. — С. 313-362.
15. Barnett L., Fujinami R. Molecular mimicry: a mechanism for autoimmune injury // Fase. BI. — 1992. — Vol. 6. — P. 840-844.
16. Bartalena L., Pinchera A., Marcocci C. Management of Graves Ophthalmopathy: reality and perspectives // Endocrine Rev. — 2000. — Vol. 21. — P. 168-198.
17. Bretz J., Baker J.R. Apoptosis and autoimmune thyroid disease: following a Trail to thyroid destruction? // Clin. Endocrinol. — 2001. — Vol. 55 (1). — P. 1-11.
18. Brix T., Kyvik K., Christensen K., Hedegus L. Evidence for a may role of heredity in Graves disease: A population-based study of two Danish twin cohorts // J. Clin. Endocrinol. Metabol. — 2001. — Vol. 86. — P. 930-934.
19. Davis T. Newer Aspects of Graves disease // Bailliere s Clin. Endocrin. Metab. — 1997. — Vol. 11. — P. 431-501.
20. Davis T., Roti E., Braverman L., De Groot L. Controversy — Thyroid Stimulating antibodies // J. Clin. Endocrinol. Metabol. — 1998. — Vol. 98. — P. 3777-3785.
21. Dayan C., Daniel G. Chronic autoimmune Thyroiditis // New. Engl. J. Med. —1996. — Vol. 335 (2). — P. 99-106.
22. De Groot L., Quintans J. The causes of autoimmune thyroid disease // Endocr. Rev. — 1989. — Vol. 10. — P. 537-562.
23. Doniach D., Botazzo G., Russel R. Goitrous autoimmune Thyroiditis (Hashimoto s Disease) // Clin. Endocrin. Metabol. — 1979. — Vol. 8 (1). — P. 63-80.
24. Freedman S., Posnett D., Tumang J. et al. A potential role for microbial superantigens in the pathogenesis of systemic autoimmunne disease // Arthritis Rheum. — 1991. — Vol. 34. — P. 468-480.
25. Nicolai T., Coombs G., McKenzie A. Lymphocytic thyroiditis with spontaneously resolving hyperthyroidism (silent thyroiditis) and subacute thyroiditis: long-term follow-up // Arch. Intern. Med. — 1981. — P. 1455-1458.
26. Othman S., Phillips D., Parkes A. et al. A long-term fellow-up of postpartum thyroiditis // Clin. Endocrinol. — 1990. — Vol. 32. — P. 559-564.
27. Pearse E., Farwell A., Braverman L. Thyroiditis // New. Engl. J. Med. —2003. — Vol. 348 (26). — P. 2646-2654.
28. Roti E., Emerson C. Postpartum thyroiditis // J. Clin. Endocrinol. Metabol. — 1992. — Vol. 74. — P. 3-5.
29. Scridama W., De Groot L. Treatment of Graves disease and course ophthalmopathy // Amer. J. Med. — 1989. — Vol. 87. — P. 70-73.
30. Tait K., Gough S. The genetics of autoimmune endocrine disease // Clin. Endocrin. — 2003. — Vol. 59 (1). — P. 1-11.
31. Tomer Y., Barbesino G., Greenberg D. et al. Mapping the major suscetibility loci for familial Graves and Hashimoto diseases: Evidence for genetic heterogenety and gene interactions // J. Clin. Endocrinol. Metabol. — 2001. — Vol. 86. — P. 626-630.
32. Tomer Y., Davis T. Infection, thyroid disease and autoimmunity // Endocr. Rev. — 1993. — Vol. 14. — P. 107-120.
33. Tsuchiga N., Willams R. Molecular mimicry — hypotesis or reality // West. J. Med. — 1992. — Vol. 157. — P. 133-138.
34. Weetman A.P. Autoimmune thyroiditis: predisposicion and pathogenesis // Clin. Endocrinol. — 1992. — Vol. 36. — P. 307-323.
35. Weetman A., Mac Gregor A. Autoimmune thyroid disease: further development in our understanding // Endocrin. Rev. — 1994. — Vol. 15. — P. 788-830.
36. Weightman D., Kendall-Taylor R. Cross-reaction of eye muscle antibodies with thyroid tissue in thyroid-associated ophthalmopathy // J. Endocrinol. — 1989. — Vol. 122. — P. 201-206.
37. Wilkin T. The primary lesion theory of autoimmunity: a speculative hypothesis // Autoimmunity. — 1990. — Vol. 7. — P. 225-235.
38. Volpe R. Suppressor T-lymphocyte dysfunction is important in the pathogenesis of autoimmune thyroid disease // Thyroid. — 1993. — Vol. 3. — P. 345-350.

/144.jpg)
/147.jpg)