Введение
Пациенты с сахарным диабетом (СД) характеризуются возрастанием риска развития сердечно-сосудистых заболеваний (ССЗ). Особенностью СД, способствующего этому, является развитие ускоренного атеросклероза (АС). Согласно эпидемиологическим расчетам, к 2020 году АС станет основной причиной смерти в мире [1].
Воспалительные процессы являются ключевыми в развитии и прогрессировании ССЗ, а повышенные уровни воспалительных маркеров, таких как С-реактивный белок и сывороточный амилоид А, являются предвестниками заболевания. Показано, что специфическое подавление этих процессов на экспериментальных моделях тормозит прогрессирование болезни, уменьшая повреждения миокарда и артерий и способствуя их заживлению [2].
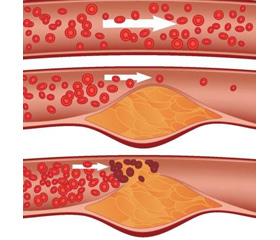
Схема образования бляшки
Ключевым событием, инициирующим атерогенез, считается эндотелиальная дисфункция (ЭД), которая может произойти в результате СД, артериальной гипертензии (АГ), в областях неламинарного сосудистого кровотока (точки ветвления и изгибы) или повышенного уровня липопротеинов низкой плотности (LDL) в плазме. Увеличение проницаемости эндотелия приводит к накоплению путем диффузии аполипопротеин В-содержащих липопротеинов (АроВ), взаимодействующих с богатым протеогликанами внеклеточным матриксом (ECM). Это взаимодействие удерживает LDL в стенке сосуда, где они подвергаются окислению с помощью активных форм кислорода (ROS). Окисленные LDL (охLDL) затем стимулируют эндотелиальные клетки (ЭК), усиливая образование молекул клеточной адгезии, белков хемотаксиса, факторов роста и подавляя продукцию окиси азота (NO). Моноциты, происходящие из костного мозга или селезенки, рекрутируются из крови в интиму, привлекаемые хемокинами (CCL2 в первую очередь), с соответствующими рецепторами CCR2, CCR5, CX3CR1, которые экспрессируются эндотелием [2, 3]. В субэндотелиальном пространстве внутренней оболочки моноциты дифференцируются в макрофаги, которые посредством рецепторов-скавенджеров (СР) захватывают oxLDL в стенку сосуда путем фагоцитоза. Поглощение модифицированных LDL (modLDL) приводит к накоплению капель холестерина в цитоплазме макрофага, создавая канонические пенистые клетки, которые типичны для ранних атеросклеротических образований. Т-клетки, в частности CD4+ Th1-клетки, также мобилизуются в ранние атеросклеротические повреждения сосудов и могут распознавать аутоантигены, в том числе oxLDL и HSP60. Th1-клетки продуцируют большое количество интерферона (IFN-γ), активирующего макрофаги, что ведет к дальнейшему синтезу цитокинов и хемокинов, активации тромбоцитов и усилению воспаления. Секреция цитокинов активированными макрофагами стимулирует пролиферацию клеток гладких мышц (ГМК) сосудов. ГМК интимы продуцируют ЕСМ, который дает начало образованию фиброзного кэпа. Такой комплекс бляшки подвержен дестабилизации, разрушению и развитию тромбоза, что может привести к острой окклюзии сосудов. Атеросклеротические бляшки при СД характеризуются усиленной кальцификацией, образованием некротических ядер, наличием рецепторов для конечных продуктов гликозилирования (RAGE), а также усиленной инфильтрацией макрофагов и Т-клеток. Наблюдается также повышенное количество бляшек с разрывами и перестроек сосудов. Эти особенности способствуют развитию более тяжелого АС [4].
Воспалительные сигнальные механизмы клеток сосудов
Воспаление — основной механизм реализации врожденного и адаптивного иммунитета. ЭК сосудов играют важную роль в инициации, амплификации и разрешении воспалительных реакций. Хроническое воспаление — причина развития различных заболеваний, таких как АС, ожирение, СД и АГ. Сигнальные пути, опосредующие воспалительные процессы в ЭК, достаточно хорошо изучены. Макромолекулярные комплексы — инфламмасомы контролируют трансдукцию провос–палительных сигналов от мембранных рецепторов к ключевым факторам транскрипции, таким как NF-κB. Инфламмасомы связаны с паттерн-распо–знающими рецепторами (RRP) — TLR, NLR (NOD-подобные рецепторы), опосредующими реакции врожденного иммунитета. ЭК сосудов образуют интерфейс между потоком крови и стенкой сосуда и выполняют ряд важных функций в поддержании гомеостаза организма [5–7]. Эндотелий не только формирует неадгезивный и высокоселективный физический барьер, контролирующий проницаемость сосудов, но и секретирует большое количество вазоактивных веществ для регулирования тонуса и ремоделирования сосудов [8]. ЭК являются важным регулятором и главной мишенью воспалительных процессов. Вследствие экспрессии ряда хемокинов и молекул клеточной адгезии эндотелий контролирует место, степень и продолжительность воспалительного процесса, обеспечивая защиту и последующее восстановление ткани после инфекции или травмы. С другой стороны, ЭК сами по себе являются объектами воспалительных реакций [6]. Воспалительная активация ЭК — одна из ключевых патофизиологических стадий при многих заболеваниях: инфекционных, аутоиммунных и нарушениях обмена веществ, таких как ожирение и СД.
Активация эндотелия и воспаление. ЭК постоянно подвергаются биологическим, химическим и механическим нагрузкам. Различные факторы в кровотоке активируют ЭК и провоцируют воспалительные реакции. Среди них — инфекционные агенты, липополисахариды (LPS), эндотоксины бактерий, нарушение кровотока [9] и TNF-α [10]. В норме ЭК поддерживают состояние покоя, выполняя антитромботическую, противовоспалительную и антипролиферативную функции. При воспалении ЭК фенотипически переходят в активированное состояние I или II типа, которое характеризуется повышенной проницаемостью, адгезией лейкоцитов и протромботической функцией. При I типе ослабляются межклеточные взаимодействия ЭК, происходит повышение проницаемости и экспорта телец Вайбеля — Паладе (экзоцитоза), а также высвобождение из гранул-хранилищ фактора VWF (von Willebrand factor) и P-селектина (CD62P), инициируя взаимодействие эндотелия с лейкоцитами и тромбоцитами. Это быстрый, но временный ответ, не зависящий от экспрессии генов de novo. Тип II активации обеспечивает более устойчивую воспалительную реакцию, стимулируя экспрессию генов различных провоспалительных цитокинов и молекул адгезии [6].
Модификация циркулирующих LDL. При АС и СД LDL сыворотки подвергаются различным химическим модификациям, нарушающим их нормальную функцию. Модификация LDL играет важную роль на начальных стадиях атерогенеза, связанных с индукцией воспаления, накоплением липидов в интиме и образованием пенистых клеток. Макрофаги отвечают за клиренс modLDL, предотвращая повреждение тканей, воспаление и нарушение обмена веществ. Они развивают сенсорный арсенал, состоящий из PRR, способных распознавать и связывать чужие или измененные объекты для их инак–тивации [7]. Липиды из удерживаемых в матриксе АpoB-липопротеинов поглощаются макрофагами с помощью ряда процессов, включая пиноцитоз и фагоцитоз агрегированных LDL. ModLDL могут захватываться макрофагами при помощи системы CР, из которых SR-A1, CD36 и LOX1 (lectin-type oxidized LDL receptor 1) играют главную роль. При СД и АС липидный баланс разрегулирован, что приводит к неспособности макрофагов полностью перерабатывать modLDL, накоплению липидов и трансформации макрофагов в пенистые клетки. СР также опосредуют различные патогенные эффекты modLDL в макрофагах посредством активации внутриклеточных сигнальных сетей. Другие PRR, такие как TLR, могут также взаимодействовать с modLDL и опосредовать их эффекты независимо или вместе с СР [2].
Изучение состава LDL плазмы крови пациентов с АС выявило различные типы модификации LDL — окисление, ацетилирование, карбамилирование, десиалирование, гликирование, приобретение отрицательного электрического заряда и образование комплексов. Последнее делает модифицированные частицы LDL особенно атерогенными. Вероятно, процесс множественной модификации LDL происходит и в крови человека, и в артериальной стенке [7, 11]. Образование гликированных LDL происходит при повышенном содержании глюкозы в крови у больных СД. Модификации, влияющие на LDL, часто сосуществуют и могут происходить одновременно, что ведет к образованию, например, гликоокисленного LDL [12]. ModLDL являются цитотоксичными и способствуют повреждению ЭК, ЭД и атерогенезу. Они усиливают окислительный стресс и снижают регенеративный потенциал эндотелия, стимулируя сенесценцию эндотелиальных клеток-предшественников в результате повреждения их ДНК [13].
Как липиды, так и белковые фрагменты LDL подвергаются модификациям, влияющим на свойства LDL и усиливающим проатерогенные эффекты. Так, изменения основного белкового компонента LDL — АpoB снижают способность LDL взаимодействовать с их рецепторами [14]. ModLDL связываются СР, которые экспрессируются на поверхности макрофагов и ЭК [15]. Инкубация LDL с культивируемыми ЭК приводит к образованию modLDL, которые распознаются и поглощаются макрофагами. Предположительно эта фракция LDL способствует накоплению внутриклеточных липидов с последующей трансформацией макрофагов, насыщенных липидами, в пенистые клетки. Показано также, что ЭК высвобождают соединения, осуществляющие конверсию LDL в oxLDL [7]. Существует множество факторов, способных опосредовать окисление LDL. ROS, продуцируемые ЭК, моноцитами, макрофагами, нейтрофилами и тучными клетками, окисляют преимущественно липидную часть LDL [16]. Металлы (Fe3+ и Cu2+) участвуют в окислении фосфолипидов LDL. Липоксигеназы катализируют окисление жирных кислот (FA) до гидропероксидных аддуктов, опосредующих окисление LDL [17]. Миелопероксидаза, специфическая для нейтрофилов, продуцирует хлорноватистую кислоту и гипотиоциановые кислоты — побочные продукты, способные присоединять кислород ко многим сайтам молекулы АpoB [18]. Частицы оxLDL содержат меньшее количество антиоксидантов по сравнению с неокисленными LDL и подвержены дальнейшему окислению, что значительно усиливает проатерогенные свойства LDL. Например, необходимая для образования пенистых клеток концентрация LDL в 40 раз превышает концентрацию oxLDL [7].
Субэндотелиальное удержание и накопление modLDL. Количественный анализ эндоцитотических везикул в люминальных ЭК при экспериментальной гиперхолестеринемии у кроликов показал значительное увеличение их числа в эндотелии областей аорты, где развивались ранние атеросклеротические поражения. Вдоль люминальной поверхности эндоцитотические везикулы могут соединяться друг с другом, образуя трансэндотелиальные каналы. И LDL, и modLDL могут проходить эндотелий через ЭК путем везикулярного транс–порта или непосредственно через эндотелиальный барьер [7]. В субэндотелиальном слое LDL взаимодействуют с белками ECM, такими как протеогликаны. Положительно заряженные аминокислотные остатки АpoB взаимодействуют с отрицательно заряженными сульфатными группами протеогликанов [19]. В АpoB имеются две протеогликан-связывающие последовательности, обогащенные основными аминокислотами и расположенные на N- и С-концевых участках. ModLDL задерживаются ECM интимы сильнее, чем нативные LDL, и атерогенность LDL и modLDL прямо зависит от их аффинности к протеогликанам стенки артерии. Удержание modLDL в субэндотелиальном слое вызывает воспаление. Так, oxLDL, связанные с ECM интимы, индуцируют воспалительную активацию ЭК, опосредованную α5β1-интегринами и связанную с продукцией эндотелием VCAM-1 [20]. Это, в свою очередь, привлекает моноциты к ЭК и способствует трансэндотелиальному траффику моноцитов в интиму, где они дифференцируются в макрофаги и интернализуют отложения modLDL, используя для клиренса СР [21].
Скавенджер-рецепторы макрофагов. СР представляют собой ряд мембранных рецепторов и растворимых изоформ, включающих внеклеточный домен. СР — поверхностные рецепторы, которые распознают многочисленные лиганды и способствуют устранению чужеродных или модифицированных объектов [22]. Рецепторы включают SR-A1, рецептор макрофагов с коллагеновой структурой SR-A2 (MARCO), CD36, SR-B1, LOX1, рецептор, экспрессируемый ЭК 1 (SREC1), SR-PSOX (CXCL16), распознающий фосфатидилсерин и охLDL [23]. Исследования in vitro показали, что деградация modLDL макрофагами опосредуется в основном SR-A1 и CD36 [78]. Дефицит этих рецепторов частично ингибировал образование пенистых клеток у ApoЕ–/– мышей, что указывает на существование других механизмов поглощения LDL [24, 25]. Кроме modLDL, СР могут связывать такие лиганды, как сложные эфиры холестерина, протеогликаны, фосфолипиды, ферритин, углеводы и даже целые апоптотические клетки [22]. В условиях воспаления поглощение липидов макрофагами может быть активировано модуляторами, такими как интерферон. IFN-γ активирует ERK, опосредующую поглощение modLDL [7, 26]. Ультраструктурный анализ макрофагов, инкубированных с modLDL in vitro, свидетельствует об их накоплении в лизосомах. Биохимические исследования показали, что после интернализации LDL деградируют в компартментах лизосом, высвобождая холестерин и жирные кислоты. Свободный холестерин переносится в эндоплазматический ретикулум (ER), где он подвергается повторной этерификации ацетил-коэнзим А холестерин-ацетилтрансферазой 1 (ACAT1) [27]. Накопление холестерина в мембранах ER приводит к его дефектной этерификации с помощью ACAT1 и дополнительно увеличивает объем депо. ER-стресс, связанный с накоплением холестерина в макрофагах, также способствует прогрессированию болезни, усиливая апоптоз в бляшках. Ускорение гибели клеток и нарушение клиренса мертвых клеток приводит к образованию в развитых атеросклеротических бляшках некротического ядра. Богатые холестерином мембранные микродомены способствуют TLR/NF-κB-опосредованной передаче провоспалительных сигналов [25].
Лизосомальная деградация oxLDL, поглощенных моноцитами, приводит к активации ERK1/2- и Akt-зависимых сигнальных путей и усиленной дифференциации моноцитов до провоспалительных макрофагов. Этот механизм включает в себя активацию транскрипционных факторов NF-κB и AP-1, которые усиливают экспрессию провоспалительных генов [28].
Связывание оxLDL с рецептором CD36 стимулирует в макрофагах провоспалительные сигнальные пути. Основным путем является активация NF-κB, который индуцирует транскрипцию многих провос–палительных генов. Макрофаги, лишенные CD36, демонстрируют низкий уровень NF-κB и снижение секреции провоспалительных цитокинов — интерлейкина 1β (IL-1β) и TNF-α. В макрофагах CD36 взаимодействует с другими рецепторами и поверх–ностными сигнальными молекулами, такими как TLR и интегрины. Так, стимуляция TLR2 приводит к усилению связывания oxLDL рецептором CD36. Соответственно, oxLDL-стимулированный CD36 способствует образованию гетеродимера TLR2/6, который активирует NF-κB, транскрипцию провоспалительных генов и высвобождение IL-1β [7]. Вместе с oxLDL и амилоидом-β CD36 участвует в индукции NLRP3-инфламмасом, опосредуя передачу oxLDL и амилоида-β в лизосомы. Амилоид-β и холестерин из деградированных oxLDL образуют кристаллы, дестабилизирующие лизосомальные мембраны, и индуцируют сборку NLRP3-инфламмасом, активация которых стимулирует опосредованный каспазой процессинг и секрецию IL-1β [29]. OxLDL ингибируют миграцию макрофагов, способствуя их накоплению в очагах поражения. При АС хроническое воспаление постоянно поддерживается из-за сохранения провоспалительных макрофагов в бляшках, где они конститутивно углубляют бесконечную воспалительную реакцию.
Молекулы адгезии, посредники ЭК-лейкоцитарных взаимодействий. Считается, что ЭК в покое не адгезивны к лейкоцитам крови. Воспалительные реакции зависят от миграции лейкоцитов через ЭК. ЭК, активированные провоспалительными факторами, экспрессируют Е- и P-селектины для привлечения и роллинга лейкоцитов по воспаленному эндотелию посредством слабого связывания между селектинами и их слабоаффинными лигандами Sialyl-LewisX (тетрасахарид, соединенный на поверхности клетки с О-гликанами) и PSGL-1 (P-selectin glycoprotein ligand 1). ЭК и резидентные макрофаги сосудов секретируют хемокины, такие как CCL2, который действует через CCR2 (рецептор C-C типа хемокина 2) и CCR4 на моноцитах и –Т-лимфоцитах, а также IL-8, который связывается с –IL-8-рецептором-α и -β на нейтрофилах [6]. Перемещаемые по эндотелию лейкоциты дополнительно активируются с помощью хемокинов, экспонированных на поверхности эндотелия, что приводит к распространению и кластеризации поверхностных интегринов, таких как LFA-1 (lymphocyte function-associated antigen-1, интегрин αLβ2) и VLA-4 (very late antigen-4; интегрин α4β1). Индуцированная воспалением экспрессия ICAM-1 и VCAM-1 на ЭК обеспечивает их взаимодействие с контр-рецепторами интегринов на лейкоцитах, прочную адгезию и трансмиграцию лейкоцитов в субэндотелиальное пространство стенки сосуда, инициируя их воспаление [30]. В процессе адгезии лейкоцитов: их захвате и активации, усилении адгезии, перемещения внутри сосудов, парацеллюлярной и трансцеллюлярной миграции, участвуют ESAM (endothelial cell-selective adhesion molecule), JAM (junctional adhesion molecule), MAC1 (macrophage antigen 1), а также MAdCAM1 (mucosal vascular addressin cell-adhesion molecule 1) [6, 31].
Трансдукция сигналов через TNFR и активация NF-κB. Экспрессия цитокинов и молекул адгезии является признаком воспалительной активации эндотелия. Ядерный фактор NF-κВ — главный ре–гулятор воспалительных реакций в ЭК, а также в других типах клеток [32, 33]. NF-κВ может быть активирован различными агонистами, такими как глюкоза, охLDL, лизофосфатидиловая кислота, липопротеины очень низкой плотности (VLDL), ангиотензин II, вирусные инфекции, нарушение кровотока. NF-κВ активирует транскрипцию генов TNF-α, IL-1, IL-8, Е-селектина, VCAM-1 и ICAM-1 путем связывания с регуляторными участками этих генов-мишеней [6].
Каскад IKK/NF-κB — основной регулятор и концентратор сигналов для различных воспалительных стимулов, которым подвергаются ЭК. TNFR-путь — прототип классического провоспалительного сигналинга. Связывание TNF-α с рецептором TNFR1 мобилизует белок TRADD (tumor necrosis factor receptor type 1-associated death domain protein), который вовлекает взаимодействующую с рецептором серин-треониновую протеинкиназу 1 (RIPK1 или RIP) и TRAF2 (TNF receptor-associated factor 2) — Е3-убиквитинлигазу и адаптерный белок [34, 35]. TRAF2 также присоединяет обладающие активностью E3-убиквитинлигазы клеточные белки-ингибиторы апоптоза (cIAP), которые соединяют воспалительные и сигнальные пути, связанные с выживанием клетки. Эти молекулы, TRADD, RIP, TRAF2 и cIAP, являются основными компонентами сигналосомы TNFR1, опосредующей трансдукцию сигналов TNF-α. TRAF2 катализирует убиквитинирование RIP, что позволяет привлечь комплекс NEMO/IKK, фосфорилирующий IKK2, с последующей активацией канонического пути NF-κB и развитием воспаления. Белки cIAP, как E3-убиквитинлигазы, не только регулируют активацию NF-κB в сигнальной цепи ниже TNFR1, но и опосредуют активацию NF-κB в ответ на сигналы врожденного иммунитета [33]. Кроме участия в контроле воспаления, трансдукция сигналов цитокинов от рецепторов мембраны к NF-κB играет важную роль в регуляции апоптоза ЭК [6].
Рецепторы, ассоциированные с врожденным иммунитетом. Собраны данные, указывающие на связь активации врожденного иммунитета с ССЗ. Так, Th1-клетки обладают выраженными проатерогенными свойствами, Treg — антиатерогенными. Эффекты Th2 и Th17 в отношении АС двойственные, нуждаются в дальнейшем изучении [36]. АС — хронический воспалительный процесс, а иммунные клетки являются важными посредниками воспаления сосудов — от образования самых ранних жировых полос до поздних стадий продвинутых бляшек. Роль врожденной иммунной системы в патогенезе ССЗ была предметом особого внимания, а воздействие на иммунную функцию в экспериментальных моделях может тормозить развитие заболевания [2].
При повреждении тканей эндотелий является одной из первых линий системы обороны организма от микроорганизмов и эндогенных веществ. В защитной реакции ЭК участвует ряд PRR, включая TLR, NLR ((NOD (nucleotide-binding oligomerization-domain)-подобные рецепторы)), RLR ((RGI-I (RNA helicases retinoic acid inducible gene-I)-подобные рецепторы)), CDS (цитозольные сенсоры ДНК) и CLR (рецепторы лектинов С-типа). PRR распознают молекулярные модели, используемые микробами, объединенные под названием PAMP (патоген–ассоциированные молекулярные паттерны), или эндогенные молекулы из раненых и стрессированных клеток — DAMP (ассоциированные с опасностью молекулярные структуры). PAMP включают различные бактериальные компоненты, такие как LPS, пептидогликан, флагеллин и вирусные двухцепочечные РНК [37]. DAMP — внутриклеточные или внеклеточные белки или их фрагменты, такие как белки теплового шока (HSP), HMGB1 (high-mobility group box 1 protein) и фрагменты гиалуронана. Среди небелковых DAMP — мочевая кислота, АТР, кристаллы холестерина и ДНК хозяина. TLR принадлежат к надсемейству IL-1/TLR рецепторов, так как содержат домен Toll/IL-1-рецептора (TIR) [6]. Все TLR являются трансмембранными белками I типа с эктодоменом, состоящим из LRR (повторы, богатые лейцином). TLR можно разделить на две группы по их клеточной локализации. TLR1, 2, 4, 5, 6, 11 расположены на клеточной мембране. TLR3, 7, 8, 9, 10, 13 локализованы во внутриклеточных везикулах, в том числе на ER, эндосомах и лизосомах [38]. TLR клеточной поверхности распознают PAMP. TLR4 является рецептором для LPS, а также активируется вирусными белками, HSP60, охLDL и фибриногеном. Эндогенные лиганды TLR2 включают фактор транскрипции HMGB1, воспалительные цитокины, фрагменты гиалуроновой кислоты и бигликан [39]. Внутриклеточные, чувствительные к нуклеиновым кислотам TLR в основном распознают микробные нуклеиновые кислоты, такие как двухцепочечную РНК (TLR3) одноцепочечную РНК (TLR7) и двухцепочечную ДНК (TLR9). Экспрессия TLR специфична для клеток разного типа. TLR4, TLR2 и его корецепторы TLR1 и TLR6 экспрессируются в сосудах человека. TLR3 преимущественно экспрессируются в аорте, в то время как TLR7 и TLR9 минимально экспрессированы в подвздошной артерии [6, 40]. Роль TLR изучалась на мышиных моделях АС, для которых воспалительная активация эндотелия является важным шагом атерогенеза [37, 41]. TLR2/4 оказывают проатерогенный эффект у мышей с гиперлипидемией. Активация TLR эндотелия способствует накоплению липидов и лейкоцитов в местах поражений. В частности, TLR2/4 в ЭК коронарной артерии человека опосредуют экспрессию BMP-2 (костного морфогенетического белка 2) — воспалительного цитокина, участвующего в кальцификации сосудов [42]. Несколько СР, таких как CD36 и рецептор oxLDL, LOX1, служат корецепторами для TLR2, модулируя его воспалительные реакции [43, 44]. Учитывая, что АС может осложняться в присутствии ряда микроорганизмов — агонистов TLR, к которым относятся такие, как цитомегаловирус (TLR3 и TLR9), вирус Коксаки B (TLR7) и вирус простого герпеса (TLR9), эти рецепторы также могут участвовать в развитии эндотелиального воспаления и АС. Передача сигналов от большинства TLR опосредуется MyD88 (myeloid differentiation primary response gene 88)-зависимым путем, приводящим к воспалительным реакциям, в то время как TLR3 активирует TRIF (TIR-domain-containing adapter-inducing interferon-β)-зависимый путь, ведущий к активации интерферона β (IFN-β). При действии PAMP или DAMP TLR димеризуется и ассоциируется с адаптером MyD88, который привлекает IRAK (IL-1 receptor-associated kinases) к TLR. IRAK активируется путем фосфорилирования и затем связывается с E3 убиквитинлигазой TRAF6. Затем комплекс IRAK-1/TRAF6 высвобождается от TLR и связывается с TGF-β-активированной киназой 1 (TAK1) и TAK1-связывающими белками — TAB1 и TAB2. TRAF6, в свою очередь, убиквитинируется и облегчает связывание TAK1 с IKK2, что приводит к активации NF-κB [6, 35].
NLR — PRR, которые в основном распознают микроорганизмы и молекулы, продуцируемые в условиях стресса. NLR можно разделить на три подсемейства: NACHT (NOD), LRR (leucine-rich repeat) и PYD (NH2-terminal pyrin domain) домен-содержащие белки (NLRP); IPAF/NAIP (ICE-protease activating factor/neuronal apoptosis inhibitory protein); NOD. NLRP насчитывает 14 членов, содержащих PYD домен [45]. IPAF и NAIP содержат CARD (caspase recruitment domain) и BIR (baculoviral inhibitory repeat) домены соответственно. Члены подсемейства NOD содержат CARD и распознают пептидогликаны, сахар, сшитые с пептидами бактериального происхождения. NLRP распознают микробные токсины, а также опасные молекулы, такие как АТР, глюкоза, холестерин. IPAF детектирует флагеллин. Распознавание этих PAMP и DAMP является триггером олигомеризации NOD и сборки NOD-сигналосом, к которым мобилизуется CARD-содержащая серин-треониновая киназа (RIP2) (44). RIP2, в свою очередь, связывает TAK1, что приводит к активации NF-κB. Олигомеризация NLRP вызывает образование инфламмасом — высокомолекулярных белковых комплексов, которые активируют провоспалительные каспазы и цитокины IL-1 и IL-18. Комплексы включают каспазу-1, каспазу-5, PYCARD/ASC (apoptosis-associated speck-like protein containing a CARD) [46]. Описано несколько видов инфламмасом, в том числе NLRP1 и NLRP3. Состав инфламмасомы зависит от типа PAMP или DAMP, инициирующих сборку. Пириновый домен NLR связывается с адаптерным белком ASC. ASC содержит PYD и CARD, с помощью которого неактивная форма каспазы-1 присоединяется к NLR, инициируя ауторасщепление и активацию прокаспазы-1. Каспаза-1 расщепляет про-IL-1β по остатку Asp116 до IL-1β и про-IL-18 до IL-18, что приводит к секреции IFN-γ, провоспалительного цитокина, играющего важную роль в реакциях врожденного иммунитета [46].
Различные типы ЭК, в том числе пупочной вены человека, аорты и микроваскулярные ЭК экспрессируют NOD1. Экспрессия NOD2 в этих клетках в обычных условиях низка, но может повышаться в присутствии провоспалительных медиаторов. В ЭК, инфицированных C.pneumoniae или L.monocytogenes, активация NF-κB и МАРК, а также секреция IL-8 зависят от NOD1 [6]. Было показано, что активация инфламмасомами IL-18 приводит к дисфункции ЭК-предшественников при системной красной волчанке, аутоиммунного заболевания, характеризующегося активацией и воспалением эндотелия [47]. Также показано, что геморрагический шок активирует NLRP3-инфламмасомы в ЭК легких [48]. В свете последствий активации инфламмасом для метаболических заболеваний и критической роли воспаления эндотелия активация инфламмасом в ЭК в ответ на различные метаболические стрессы, а также регулирующие их механизмы остаются важными вопросами для дальнейшего исследования.
Роль макрофагов в воспалении
АС — хроническое воспалительное состояние, в котором макрофаги играют разные и важные роли. Фагоцитарные провоспалительные клетки проникают в растущие атеросклеротические поражения, активно участвуя в накоплении холестерина. Более того, макрофаги способствуют образованию сложных и нестабильных бляшек, поддерживая провоспалительную микросреду. В то же время противовоспалительные макрофаги способствуют восстановлению тканей, ремоделированию и стабилизации бляшек. Поэтому макрофаги представляют собой привлекательную мишень для новых методов антиатеросклеротической терапии, которые могут быть направлены на снижение рекрутирования моноцитов в места поражения, ингибирование провоспалительных макрофагов, стимулирование противовоспалительных реакций и оттока холестерина [25].
Макрофаги играют важнейшую роль на всех стадиях развития АС [49]. Считается, что моноциты и макрофаги образуют неразрывную систему, которой отводится центральное место во врожденной иммунной реакции. В сайте поражения моноциты дифференцируются в макрофаги, которые активно участвуют в иммунном ответе, высвобождая провоспалительные факторы и захватывая патогены и поврежденные клетки посредством фагоцитоза. С другой стороны, макрофаги также отвечают за разрешение воспалительного ответа и ремоделирование тканей. Согласно классической схеме, макрофаги — терминально дифференцированные клетки, которые постоянно обновляются моноцитами, выводимыми из циркуляции. Новые исследования показали, что онтогенез тканевых макрофагов более сложный, и большая часть их происходит от резидентных клеток-предшественников [50]. Субэндотелиальный слой интимы артериальной стенки человека содержит популяцию плюрипотентных перицитоподобных клеток, которые могут дифференцироваться в различные типы клеток, включая фагоциты с маркером макрофагов CD68 [51]. Показано также, что ГМК, нагруженные холестерином, теряли маркеры дифференцировки и экспрессировали маркеры макрофагов. Эксперименты in vivo согласуются с этими данными: до 40 % клеток, классифицированных как макрофаги бляшек, происходили из ГМК сосудов [2, 52].
Циркулирующие моноциты можно разделить на несколько различных типов, основанных на экс–прессии поверхностных молекул и рецепторов хемокинов [4]. Наиболее распространены классические моноциты, положительные по антигенам CD14 и отрицательные по CD16. Как и мышиные провоспалительные (LY6Chi) моноциты, эти клетки экспрессируют CCR2. Моноциты, положительные по CD16, можно разделить на 2 подмножества: CD14+СD16++ (неклассический) и CD14++CD16+ (промежуточный). Хотя оба подкласса могут секретировать провоспалительные факторы, их функции в организме различны. Неклассические моноциты, патрулирующие ткани, селективно продуцируют провоспалительные факторы в ответ на вирусы, комплексы, содержащие нуклеиновые кислоты, и, вероятно, ответственны за локальный иммунный ответ. Промежуточные моноциты в ответ на стимуляцию способны продуцировать большое количество провоспалительных молекул, таких как TNF [25].
Во время гематопоэза дифференциация моноцитов в макрофаги инициируется в основном двумя факторами роста — GM-CSF и M-SCF (гранулоцитарно-макрофагальный и макрофагальный колониестимулирующий факторы). Дифференциация циркулирующих моноцитов может быть вызвана различными стимулами, например в ответ на инфекцию или асептическое воспаление, играющее важную роль в патогенезе АС. На ранних стадиях развития АС моноциты могут рекрутироваться в жирные полоски и проникать в артериальную стенку при повышении проницаемости эндотелия, связанном с локальной ЭД. У мышей как провоспалительные, так и патрулирующие моноциты могут мобилизовываться в растущие атеросклеротические бляшки с помощью катализируемого P- и E-селектином роллинга с последующей ICAM1- и VCAM1-зависимой адгезией [25]. Провоспалительная миграция моноцитов к артериальной стенке опосредуется сигналингом CCR2, CCR5 и CX3CR1, ингибирование которого в мышиной модели АС ApoЕ–/– предот–вращало рекрутирование моноцитов и уменьшало размер поражений [53]. Моноциты, дифференцирующиеся в макрофаги, демонстрируют ряд морфологических и структурных изменений, включая увеличение размеров и количества органелл, усиление метаболизма и экспрессии поверхностных рецепторов, изменение чувствительности к сигнальным молекулам. В них также увеличивается активность лизосомальных ферментов, которая подготавливает клетки к активному фагоцитозу и перевариванию поглощенного материала [54].
Гетерогенность макрофагов. Классическая модель активации макрофагов определила два основных фенотипа: провоспалительный M1 и альтернативный M2 [4]. Дифференцировка M1-макрофагов происходит в ответ на TLR- и IFN-γ-сигналинг и может быть вызвана PAMP, LPS и LDL. Эти клетки секретируют провоспалительные факторы, такие как TNF-α, IL-1β, IL-12 и IL-23 и хемокины CXCL9, CXCL10 и CXCL11. М1-макрофаги продуцируют больше ROS и NO, которые способствуют развитию воспалительной реакции [55]. Противовоспалительные M2-макрофаги индуцируются в ответ на цитокины Th2-типа — IL-4 и IL-13 и секретируют противовоспалительные факторы, такие как агонист рецептора IL-1 и IL-10. Макрофаги, соответствующие типам M1 и M2, содержатся в атеросклеротических поражениях. M1-макрофагами обогащены прогрессирующие, а макрофагами M2 — регрессирующие бляшки, где они участвуют в восстановлении и ремоделировании тканей [55]. Недавно были выделены дополнительные классы макрофагов [56]. Тип M2 разделили на несколько подгрупп в зависимости от стимулов активации и характера экспрессии белков. M2a-макрофаги, индуцированные IL-4 и IL-13, экспрессируют высокие уровни CD206 — агониста рецептора IL-1 (IL1RN). M2b-макрофаги могут индуцироваться иммунными комплексами и лигандами IL-1R через TLR-сигналинг. Они продуцируют как противовоспалительные (IL-10), так и провоспалительные (IL-6, TNF-α) цитокины. M2c-макрофаги, которые индуцируются IL-10, TGF-β и глюкокортикостероидами, обладают противовоспалительными свойствами и продуцируют пентраксин-3, TGF-β и IL-10. Они экспрессируют рецепторную киназу Mer (MERTK) и ответственны за утилизацию апоптотических клеток. Макрофаги M2d, дифференцирующиеся в ответ на TLR-сигналинг через аденозин-A2A-рецептор, обладают ангиогенными свойствами и могут играть роль в прогрессировании опухолей и развитии атеросклеротических бляшек [25].
Роль различных типов макрофагов при АС. Как указывалось выше, кроме значительных количеств провоспалительных макрофагов (М1), в бляшках также присутствуют макрофаги М2 [55, 57]. Развитие бляшки связано с увеличением обеих популяций, но клетки, экспрессирующие провоспалительные маркеры, преимущественно распределены в плечевых областях бляшки, более восприимчивых к разрыву, а клетки с маркерами альтернативной активации расположены в адвентиции. Показано, что противовоспалительные макрофаги присутствуют в стабильных областях бляшек и более устойчивы к образованию пенистых клеток [25].
Кроме типичных M1- и M2-макрофагов атеросклеротические поражения человека содержат специфические фенотипы макрофагов с про- или антиатерогенными свойствами. Например, CD163-экспрессирующие макрофаги могут быть найдены в геморрагических бляшках [58]. Эти клетки ответственны за клиренс гемоглобина и играют защитную роль при АС. Другим атеропротективным подтипом макрофагов является Mhem. Эти клетки также экс–прессируют CD163, а также гем-зависимый транс–крипционный фактор (ATF1), который индуцирует экспрессию гем-оксигеназы 1 и LXR-β (liver X receptor). Mhem-макрофаги участвуют в клиренсе гемоглобина через фагоцитоз эритроцитов и увеличивают отток холестерина путем экспрессии LXR-β-зависимых генов — LXR-α и АТР-связывающего кассетного транспортера 1 (ABCA1) [59]. Эти клетки продуцируют IL-10 и АроE. Недавно описанные M4-макрофаги могут иметь проатерогенные свойства и участвовать в формировании нестабильных бляшек, продуцируя MMP12 и способствуя дестабилизации фиброзного кэпа бляшки [55].
Влияние липидов на активацию макрофагов. LDL — основной источник липидов при развитии АС. Опыты in vitro показали, что накопление внутриклеточного холестерина происходит не от нативных, а от атерогенных modLDL. В отличие от нативных LDL modLDL интернализуются главным образом через нерегулируемый фагоцитоз. Макрофаги с их хорошо развитым фагоцитарным аппаратом играют ключевую роль в этом процессе [55]. Показано, что как нативные LDL, так и modLDL способствуют провоспалительной поляризации макрофагов. Макрофаги распознают modLDL посредством TLR и СР. Так, рецептор CD36 может распознавать охLDL и, взаимодействуя с TLR, инициировать воспалительный сигналинг [44], что способствует поляризации макрофагов к фенотипу М1. Активация TLR сопровождается повышением активности протеинкиназ C и Syk, NADPH-оксидазы 2 (gp91/Nox2) и увеличением продукции ROS. В результате макрофаги секретируют провоспалительные цитокины, включая IL-1β, и CCL5. ОхLDL могут индуцировать воспалительный процесс непосредственно через CD36-сигналинг [60] и сдвигать фенотип макрофагов М2 к М1, изменяя экспрессию про- и противовоспалительных генов [25].
Активация инфламмасом в макрофагах может быть результатом фагоцитоза кристаллов холестерина, которые повреждают лизосомальную систему [61]. Эфиры холестерина, присутствующие в липидном ядре бляшки, могут стимулировать макрофаги, способствовать воспалению и образованию пенистых клеток. Действие холестериновых эфиров может передаваться различными сигнальными путями. Так, 7-кетохолестерил-9-карбоксинонаноат активирует NF-κB и холестерил-линолеат-MAPК [62]. Другим производным холестерина, присутствующим в атеросклеротических бляшках, является оксистерол. В макрофагах он может индуцировать экспрессию провоспалительного CCL2 и рецепторов CD36. Экспрессия CD36 также стимулируется окисленными эфирами холестерина. Этот рецептор играет важную роль в атерогенезе, поскольку его уменьшение посредством стимуляции αМβ2-интегринов подавляет образование воспалительных макрофагов и пенистых клеток [25].
В закисленном микроокружении бляшек фосфолипаза может гидролизовать LDL, что приводит к высвобождению свободных фосфолипидов и FA, способствующих накоплению липидов в артериальной стенке и прогрессии бляшек. Было показано, что LDL, обработанные фосфолипазой А2, увеличивают секрецию макрофагами TNF-α, IL-6 и стимулируют образование пенистых клеток. Провоспалительный сигналинг фосфолипидов и FA опосредуется G-белок-связанным рецептором G2A, который играет важную роль в патогенезе, поскольку его дефицит приводит к развитию АС и приобретению макрофагами фенотипа M1 [25]. Насыщенные FA способствуют сдвигу фенотипа макрофагов к М1 посредством TLR/NF-κB-сигналинга [63].
Полиненасыщенные жирные кислоты (PUFA) обладают защитными свойствами при АС, что частично объясняется их противовоспалительным действием на макрофаги. Конъюгированная линолевая кислота уменьшала экспрессию генов, участвующих в воспалительных процессах (NF-κB, CCL2, MMP9, фосфолипазы-2 и циклооксигеназы-2) в макрофагах через PPARγ (peroxisome proliferator-activated receptor γ), и подавляла прогрессию АС у мышей. PUFA также могут противодействовать проатеросклеротическим эффектам насыщенных FA, например индуцированной пальмитатом экс–прессии LOX1 и белка, связывающего FA [64]. Эйкозапентаеновая и дигидроаскорбиновая кислоты оказывают защитные эффекты при АС, уменьшая провоспалительную активность и улучшая функциональность макрофагов. Нитрожирные кислоты (NFA) образуются при взаимодействии активных форм азота с FA при окислительном стрессе. Показано, что NFA обладает противовоспалительными и атеропротективными свойствами, опосредуемыми Nrf2 и PPARγ. Ослабление АС и стабилизация бляшек в результате повышенного депонирования коллагена наблюдалось у мышей ApoЕ–/–, получавших NFA [25].
HDL характеризуются атеропротекторными функциями, стимулируя отток холестерина и катаболизм [4]. У больных АС наблюдается снижение уровней HDL против LDL. Нормализация содержания HDL в сыворотке у мышей с АС приводила к уменьшению в очагах поражения количества макрофагов М1 и увеличению M2 с маркерами CD163, Arg-1 и фактором транскрипции FIZZ1. Экспрессия Arg-1 и FIZZ1 зависела от STAT6 [65]. HDL ингибируют провоспалительную поляризацию макрофагов, судя по маркерам TNF-α, IL-6 и CCL2, а также маркерам клеточной поверхности, но не стимулируют активацию макрофагов противовоспалительного фенотипа [4].
Формирование пенистых клеток. LDL, циркулирующие в крови здоровых лиц, обычно не вызывает накопления липидов в культивируемых макрофагах, тогда как LDL пациентов с АС в большинстве случаев является индуктором клеточного липидоза. Таким образом, LDL пациентов с АС в отличие от LDL здоровых лиц является атерогенным. При добавлении к первичной культуре макрофагов LDL, выделенные из крови пациентов с АС, индуцируют повышенную экспрессию TNF-α и противовоспалительного хемокина CCL18. В то же время нативные LDL от здоровых лиц не влияли на экспрессию генов при добавлении к макрофагам. Следовательно, атерогенные modLDL вызывают активацию про- и противовоспалительных макрофагов. Это важный факт, учитывая значительную роль врожденного иммунитета и хронического воспаления при возникновении и развитии АС. Полученные данные согласуются с результатами исследований in situ, которые показали усиление экспрессии про- и противовоспалительных цитокинов при АС [66].
Для оценки влияния индуцированного modLDL накопления холестерина на экспрессию генов в макрофагах было проведено исследование транскриптома макрофагов, инкубированных с окисленными, ацетилированными и десиалированными LDL. Добавление modLDL приводило к изменениям в активности сотен генов, 26 из которых связаны с функцией врожденного иммунитета. Следовательно, LDL-индуцированное накопление холестерина в макрофагах вызывает иммунный ответ [25].
Повышение оттока липидов может стать новым терапевтическим подходом для лечения АС. Несколько белков облегчают отток липидов из макрофагов, включая ABCA1 и ABCG1, которые опосредуют переход липидов к HDL-частицам и активируются в ответ на рост уровня холестерина в клетках, детектируемого сенсорами LXR. Их активация оказывает противовоспалительное действие. Так, активацию LXR в мышиных макрофагах с дефицитом ABCA1/G1 связывают с сильным антиатеросклеротическим эффектом и уменьшением площади поражения за счет уменьшения воспаления [67]. В настоящее время изучается возможность активации LXR для лечения АС. Другим механизмом клиренса холестерина в клетках является особый тип аутофагии — липофагия. Исследования на модели мышиного АС показали защитную роль аутофагии через регуляцию воспаления и гибели клеток в атеросклеротических бляшках [68].
Считается, что неоваскуляризация из vasa vasorum способствует кровоизлияниям внутри бляшек, что не только ускоряет их увеличение и воспаление, но также располагает к разрыву. Ангиогенез стимулируется VEGF — мишенью для индуцируемого гипоксией фактора (HIF1α), особенно в пределах больших, богатых липидами некротических ядер, присутствующих в развитой атероме. В исследованиях in vitro гипоксия и экспрессия HIF1α изменяли состояние липидов и подавляли отток холестерина через транспортеры ABCA1 в макрофагах. Более того, эксперименты in vitro и ex vivo показали, что гипоксия и HIF1α в связаны с усилением поглощения глюкозы, метаболической активности и поляризации макрофагов в атероме человека [2].
Взаимодействие макрофагов и нейтрофилов. Ранние поражения характеризуются инфильтрацией нейтрофилов и загруженных липидами макрофагов. При развитии поражений нейтрофилы и макрофаги продолжают накапливаться и рекрутируют провоспалительные IL-17-продуцирующие Т-клетки. Усиливается пролиферация ГМК и изменение образования матрикса [69]. Нейтрофилы стимулируют макрофаги для провоспалительных реакций в атеросклеротических бляшках. Эта инициация опосредуется внеклеточными нейтрофильными ловушками (NET) — трехмерными сетями, основу которых составляет ДНК, связанная с цитотоксическими гистонами, и белками гранул, которые высвобождаются активированными нейтрофилами. Этот процесс, называемый нетозом, является скоординированным многоступенчатым процессом: цитруллинация гистонов, деконденсация хроматина, миграция эластазы и других ферментов гранул в ядро, дезинтеграция ядерной мембраны и высвобождение ДНК, гистонов и белков гранул во внеклеточное пространство. Высвобождение NET инициирует образование в макрофагах про-IL-1β, которые расщепляются каспазой-1 (секретируется макрофагами в ответ на активацию NLRP3-инфламмасом) до зрелых провоспалительных IL-1β [70].
Нейтрофилы от больных СД 1-го и 2-го типа, а также диабетических мышей инициируют образование NET и характеризуются повышенной экспрессией пептидиларгининдеиминазы 4 (PAD4) — фермента, критического для опосредованной цитруллинацией гистонов деконденсации хроматина и образования NET [71]. Усиление транскрипции PAD4 управляется NFκB, хронически активированным при СД. Индуцированные гипергликемией ROS, вероятно, активируют также PAD4, увеличивая внутриклеточную концентрацию Ca2+. Другие последствия образования ROS, такие как активация PKC, увеличивают уровни TNF-α, подготавливающего нетоз [69]. Разрешение воспаления, вызванного инфильтрацией нейтрофилов и макрофагов, включает в себя переход от синтеза из арахидоновой кислоты простагландинов и лейкотриенов к синтезу липоксинов, которые останавливают рекрутирование нейтрофилов [35]. Одновременно увеличение продукции резолвинов и протектинов из омега-3-PUFA вызывает апоптоз нейтрофилов. Фагоцитоз нейтрофилов макрофагами вызывает их переход к фенотипу М2-макрофагов, которые выделяют противовоспалительные и репаративные цитокины [69].
Активация в макрофагах NLRP3-инфламмасом при СД и АС. Повышенная экспрессия компонентов инфламмасом была обнаружена в моноцитах у пациентов с СД 2-го типа до лечения. Наряду с этим наблюдалась усиленная активация инфламмасом. Соответственно в сыворотке таких пациентов были значительно более высокие уровни провоспалительных цитокинов IL-1β и IL-18, чем у здоровых людей [72]. NF-κB, который хронически активен в мононуклеарах у пациентов с диабетом и в ЭК сосудов диабетических крыс, способствует транс–крипции NLRP3, pro-IL-1β и pro-IL-18 [73]. Эти белки остаются в цитоплазме в неактивных формах. Второй сигнал активирует NLRP3-инфламмасомы, способствуя олигомеризации неактивного NLRP3, ASС и прокаспазы-1. Многие, но не все активаторы NLRP3 сходятся на избыточной продукции ROS. ROS-индуцированное снижение внутриклеточных уровней NAD+ уменьшает активность деацетилазы SIRT2, вызывая накопление ацетилированного α-тубулина, регулирующего перенос митохондрий и способствующего эффективному взаимодействию между адаптерным белком ASС и NLRP3 [74]. ROS также активирует NLRP3-инфламмасомы, открывая кальциевые каналы клеточной мембраны TRPM2, увеличивая приток Ca2+. Митохондриальный мембранный фосфолипид кардиолипин активирует NLRP3-инфламмасомы после транслокации (возможно, в результате ROS-индуцированного ремоделирования кардиолипина) на внешнюю митохондриальную мембрану, где он связывается с NLRP3 [75]. Активация NLRP3 в артериях больных СД может быть связана с увеличением на клеточной поверхности количества CD36, индуцированного инсулинорезистентностью (ИР), что облегчает интернализацию охLDL и внутриклеточную конверсию охLDL в кристаллы холестерина [69].
В свиной модели диабетического АС процессинг SREBP-1 и SREBP-2 и экспрессия их генов-мишеней усиливались в ЭК и инфильтрирующихся макрофагах как в первичных жирных полосках, так и в продвинутых бляшках с фиброзными крышками, некротическими и холестериновыми ядрами. Одновременно уменьшалась активность SIRT1 и AMPK. Увеличение содержания SREBP-1a в макрофагах при АС диабетических свиней непосредственно регулирует транскрипцию NLRP3 [76]. Возросшее количество NLRP3 обнаружили как в макрофагах развитых бляшек, так и в ЭК и ГМК. Изменения, обнаруженные у свиней, также выявлены в образцах коронарного АС у пациентов с СД.
ИР повышает окисление FA в артериальных ЭК. На двух инсулинорезистентных недиабетических моделях животных показано, что ингибирование высвобождения FFA из адипоцитов либо их окис–ления в артериальном эндотелии предотвращало увеличение продукции ROS и его патологических эффектов. В артериальных ЭК индуцированное FFA увеличение ROS активирует те же пути, которые наблюдаются при гипергликемии: AGE, PKC, гексозаминовый путь и NFκB. Избыточное количество супероксидов также активирует множество провоспалительных сигналов, принимающих участие в индуцированном гипергликемией повреждении сосудов, а также инактивирует два важных антиатерогенных фермента: простациклин-синтазу и eNOS [77].
Аутоиммунность АС
Клетки как врожденной (макрофаги), так и адаптивной (Т- и В-лимфоциты, дендритные клетки) иммунных систем могут модулировать локальную воспалительную среду. В зависимости от среды –Т-лимфоциты могут секретировать провоспалительные Th1-цитокины, такие как IL-1, IL-6 и TNF, или Th2-цитокины — IL-4, IL-10 и IL-13, которые способствуют разрешению воспаления. IL-1- и TNF-сигналинг в основном опосредуются р38MAPK/NF-κB. Активация сигналинга IL-6 через трансдуцирующий сигнал белок gp130, который активирует JAK1 и STAT1/3, приводит к активации ЭК и макрофагов с образованием молекул адгезии и хемокинов [78].
В настоящее время собраны доказательства в пользу гипотезы о том, что АС является аутоиммунным заболеванием, обусловленным накоплением и модификацией липопротеинов в стенке сосуда и их детектированием специфическими Т-клетками и антителами.
Следующие данные подтверждают эту гипотезу:
1. Т-клетки инфильтруются в стенки аорты, накапливаются и демонстрируют ограниченный репертуар рецепторов Т-клеток.
2. Активация Т-клеток в бляшке поддерживается взаимодействием с резидентными антигенпредставляющими клетками и требует презентации специфических антигенов.
3. Аутоантитела к липидным антигенам и их белковым группам являются атеропротектными и связаны с ослаблением АС.
4. Развитие АС мыши можно модулировать иммунизацией против некоторых известных антигенов. Эти данные создают теоретическую основу для понимания АС как аутоиммунного заболевания [79, 80].
Выводы
Доказано, что в основе патогенеза СД и вызванного диабетом атеросклероза лежат хронические воспалительные процессы. В воспалении участвуют различные типы клеток, рецепторов, протеинкиназ/фосфатаз, транскрипционных факторов, цитокинов/хемокинов и множество других биохимических соединений, взаимодействие которых в пространстве и времени определяет направленность процессов.
Ключевым фактором, интегрирующим в клетке поступающие извне провоспалительные сигналы и реализующим их эффект, является NF-κB (рис. 1). Изучение тонких механизмов воспаления и его разрешения, а также поиск способов воздействия на эти механизмы являются необходимыми условиями понимания патогенеза АС и разработки новых подходов для его лечения.
Конфликт интересов. Авторы заявляют об отсутствии какого-либо конфликта интересов при подготовке данной статьи.
Список литературы
1. Husain K., Hernandez W., Ansari R.A., Ferder L. Inflammation, oxidative stress and renin angiotensin system in athe–rosclerosis // World J. Biol. Chem. — 2015. — Vol. 6, № 3. — Р. 209-217.
2. Ruparelia N., Chai J.T., Fisher E.A., Choudhury R.P. Inflammatory processes in cardiovascular disease: a route to targeted therapies // Nat. Rev. Cardiol. — 2017. — Vol. 14, № 3. — P. 133-144.
3. White G.E., Iqbal A.J., Greaves D.R. CC chemokine receptors and chronic inflammation — therapeutic opportunities and pharmacological challenges // Pharmacol. Rev. — 2013. — Vol. 65, № 1. — P. 47-89.
4. Sokolova L.K., Pushkarev V.M., Pushkarev V.V., Tronko M.D. Diabetes and atherosclerosis. Cellular mechanisms of the pathogenesis (review) // Endokrynologia. — 2017. — Vol. 22, № 2. — P. 127-138.
5. Aird W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms // Circ. Res. — 2007. — Vol. 100. — P. 158-173.
6. Xiao L., Liu Y., Wang N. New paradigms in inflammatory signaling in vascular endothelial cells // Am. J. Physiol. Heart Circ. Physiol. — 2014. — Vol. 306, № 3. — P. 317-325.
7. Chistiakov D.A., Melnichenko A.A., Orekhov A.N., Bobryshev Y.V. How do macrophages sense modified low-density lipoproteins? // Int. J. Cardiol. — 2017. — Vol. 230. — P. 232-240.
8. Tousoulis D., Kampoli A.M., Papageorgiou N., Androulakis E., Antoniades C., Toutouzas K., Stefanadis C. Pathophysio–logy of atherosclerosis: the role of inflammation // Curr. Pharm. Des. — 2011. — Vol. 17. — P. 4089-4110.
9. Dunn J., Simmons R., Thabet S., Jo H. The role of epigenetics in the endothelial cell shear stress response and atherosclerosis // Int. J. Biochem. Cell Biol. — 2015. — Vol. 67. — P. 167-176.
10. Brevetti G., Giugliano G., Brevetti L., Hiatt W.R. Inflammation in peripheral artery disease // Circulation. — 2010. — Vol. 122. — P. 1862-1875.
11. Ivanova E.A., Bobryshev Y.V., Orekhov A.N. LDL electronegativity index: a potential novel index for predicting cardiovascular disease // Vasc. Health Risk Manag. — 2015. — Vol. 11. — P. 525-532.
12. Soran H., Durrington P.N. Susceptibility of LDL and its subfractions to glycation // Curr. Opin. Lipidol. — 2011. — Vol. 22. — P. 254-261.
13. Carracedo J., Merino A., Briceño C., Soriano S., Buendía P., Calleros L., Rodriguez M., Martín-Malo A., Aljama P., Ramírez R. Carbamylated low-density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells // FASEB J. — 2011. — Vol. 25. — P. 1314-1322.
14. Alique M., Luna C., Carracedo J., Ramírez R. LDL biochemical modifications: a link between atherosclerosis and aging // Food Nutr. Res. — 2015. — Vol. 59. — 29240. — doi: 10.3402/fnr.v59.29240.
15. Orekhov A.N., Bobryshev Y.V., Sobenin I.A., Melnichenko A.A., Chistiakov D.A. Modified low density lipoprotein and lipoprotein-containing circulating immune complexes as diagnostic and prognostic biomarkers of atherosclerosis and type 1 diabetes macrovascular disease // Int. J. Mol. Sci. — 2014. — Vol. 15. — P. 12807-12841.
16. Yoshida H., Kisugi R. Mechanisms of LDL oxidation // Clin. Chim. Acta. — 2010. — Vol. 411. — P. 1875-1882.
17. Wittwer J., Hersberger M. The two faces of the 15-lipoxygenase in atherosclerosis // Prostaglandins Leukot. Essent. Fat. Acids. — 2007. — Vol. 77. — P. 67-77.
18. Delporte C., Boudjeltia K.Z., Noyon C. et al. Impact of myeloperoxidase-LDL interactions on enzyme activity and subsequent posttranslational oxidative modifications of apoB-100 // J. Lipid. Res. — 2014. — Vol. 55. — P. 747-757.
19. Fogelstrand P., Borén J. Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis // Nutr. Metab. Cardiovasc. — 2012. — Vol. 22. — P. 1-7.
20. Yurdagul A.Jr., Green J., Albert P. et al. α5β1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis // Arterioscler. Thromb. Vasc. Biol. — 2014. — Vol. 34. — P. 1362-1373.
21. Zani I.A., Stephen S.L., Mughal N.A., Russell D., Ho–mer-Vanniasinkam S., Wheatcroft S.B., Ponnambalam S. Sca–venger receptor structure and function in health and disease // Cells. — 2015. — Vol. 4. — P. 178-201.
22. Prabhudas M., Bowdish D., Drickamer K. et al. Standardizing scavenger receptor nomenclature // J. Immunol. — 2014. — Vol. 192. — P. 1997-2006.
23. Moore K.J., Sheedy F.J., Fisher E.A. Macrophages in atherosclerosis: a dynamic balance // Nat. Rev. Immunol. — 2013. — Vol. 13, № 10. — P. 709-721.
24. Manning-Tobin J.J., Moore K.J., Seimon T.A. et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice // Arterioscler. Thromb. Vasc. Biol. — 2009. — Vol. 29, № 1. — P. 19-26.
25. Bobryshev Y.V., Ivanova E.A., Chistiakov D.A. et al. Macrophages and their role in atherosclerosis: pathophysio–logy and transcriptome analysis // Biomed. Res. Int. — 2016. — 2016. — P. 1-13. — doi: 10.1155/2016/9582430.
26. Li N., McLaren J.E., Michael D.R. et al. ERK is integral to the IFN-γ-mediated activation of STAT1, the expression of key genes implicated in atherosclerosis, and the uptake of modified lipoproteins by human macrophages // J. Immunol. — 2010. — Vol. 185. — P. 3041-3048.
27. Chistiakov D.A., Bobryshev Y.V., Orekhov A.N. Macrophage-mediated cholesterol handling in atherosclerosis // J. Cell. Mol. Med. — 2016. — Vol. 20, № 1. — P. 17-28.
28. Radhika A., Sudhakaran P.R. Upregulation of macro–phage-specific functions by oxidized LDL: lysosomal degradation-dependent and -independent pathways // Mol. Cell. Biochem. — 2013. — Vol. 372. — P. 181-190.
29. Sheedy F.J., Grebe A., Rayner K.J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation // Nat. Immunol. — 2013. — Vol. 14. — P. 812-820.
30. Cook-Mills J.M., Deem T.L. Active participation of endothelial cells in inflammation // J. Leukoc. Biol. — 2005. — Vol. 77. — P. 487-495.
31. Ley K., Laudanna C., Cybulsky M.I., Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated // Nat. Rev. Immunol. — 2007. — Vol. 7. — P. 678-689.
32. Rahman A., Fazal F. Blocking NF-kappaB: an inflam–matory issue // Proc. Am. Thorac. Soc. — 2011. — Vol. 8. — P. 497-503.
33. Пушкарьов В.М., Ковзун О.І., Пушкарьов В.В., Гуда Б.Б., Тронько М.Д. Хронічне запалення і рак. Значення ядерного фактора NF-кB // Журнал АМНУ. — 2015. — Т. 21, № 3–4. — С. 287-298.
34. Silke J. The regulation of TNF signalling: what a tangled web we weave // Curr. Opin. Immunol. — 2011. — Vol. 23. — P. 620-626.
35. Пушкарьов В.М., Соколова Л.К., Ковзун О.І., Пушкарьов В.В., Тронько М.Д. Участь ядерного фактора NF-κB у патогенезі діабету 1-го типу // Ендокринологія. — 2016. — Т. 21, № 3. — C. 225-238.
36. Taleb S. Inflammation in atherosclerosis // Arch. Cardiovasc. Dis. — 2016. — Vol. 109, № 12. — P. 708-715.
37. Tobias P.S., Curtiss L.K. Toll-like receptors in atherosclerosis // Biochem. Soc. Trans. — 2007. — Vol. 35. — P. 1453-1455.
38. Gay N.J., Gangloff M. Structure and function of Toll receptors and their ligands // Annu. Rev. Biochem. — 2007. — Vol. 76. — P. 141-165.
39. Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors // Nat. Immunol. — 2010. — Vol. 11. — P. 373-384.
40. Pryshchep O., Ma-Krupa W., Younge B.R. et al. Vessel-specific Toll-like receptor profiles in human medium and large arteries // Circulation. — 2008. — Vol. 118. — P. 1276-1284.
41. Curtiss L.K., Tobias P.S. Emerging role of Toll-like receptors in atherosclerosis // J. Lipid. Res. — 2009. — Vol. 50. — P. 340-345.
42. Su X., Ao L., Shi Y., Johnson T.R. et al. Oxidized low density lipoprotein induces bone morphogenetic protein-2 in coro–nary artery endothelial cells via Toll-like receptors 2 and 4 // J. Biol. Chem. — 2011. — Vol. 286. — P. 12213-12220.
43. Seimon T.A., Nadolski M.J., Liao X. et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress // Cell. Metab. — 2010. — Vol. 12. — P. 467-482.
44. Stewart C.R., Stuart L.M., Wilkinson K. et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer // Nat. Immunol. — 2010. — Vol. 11, № 2. — P. 155-161.
45. Ting J.P., Lovering R.C., Alnemri E.S. et al. The NLR gene family: a standard nomenclature // Immunity. — 2008. — Vol. 28. — P. 285-287.
46. Schroder K., Tschopp J. The inflammasomes // Cell. — 2010. — Vol. 140. — P. 821-832.
47. Kahlenberg J.M., Thacker S.G., Berthier C.C. et al. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus // J. Immunol. — 2011. — Vol. 187. — P. 6143-6156.
48. Xiang M., Shi X., Li Y. et al. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells // J. Immunol. — 2011. — Vol. 187. — P. 4809-4817.
49. Moore K.J., Tabas I. Macrophages in the pathogenesis of atherosclerosis // Cell. — 2011. — Vol. 145, № 3. — P. 341-355.
50. Ginhoux F., Jung S. Monocytes and macrophages: deve–lopmental pathways and tissue homeostasis // Nat. Rev. Immunol. — 2014. — Vol. 14, № 6. — P. 392-404.
51. Orekhov A.N., Bobryshev Y.V., Chistiakov D.A. The complexity of cell composition of the intima of large arteries: focus on pericyte-like cells // Cardiovasc. Res. — 2014. — Vol. 103, № 4. — P. 438-451.
52. Feil S., Fehrenbacher B., Lukowski R. et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis // Circ. Res. — 2014. — Vol. 115. — P. 662-667.
53. Combadiere C., Potteaux S., Rodero M. et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice // Circulation. — 2008. — Vol. 117, № 13. — P. 1649-1657.
54. Novoselov V.V., Sazonova M.A., Ivanova E.A., Orekhov A.N. Study of the activated macrophage transcriptome // Exp. Mol. Pathol. — 2015. — Vol. 99, № 3. — P. 575-580.
55. Chistiakov D.A., Bobryshev Y.V., Nikiforov N.G. et al. Macrophage phenotypic plasticity in atherosclerosis: the associa–ted features and the peculiarities of the expression of inflammatory genes // Int. J. Cardiol. — 2015. — Vol. 184, № 1. — P. 436-445.
56. Murray P., Allen J., Biswas S. et al. Macrophage activation and polarization: nomenclature and experimental guidelines // Immunity. — 2014. — Vol. 41, № 1. — P. 14-20.
57. Cochain C., Zernecke A. Macrophages in vascular inflammation and atherosclerosis // Pflugers. Arch. — 2017. — Vol. 469, № 3–4. — P. 485-499.
58. Boyle J.J., Harrington H.A., Piper E. et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype // Am. J. Pathol. — 2009. — Vol. 174, № 3. — P. 1097-1108.
59. Boyle J.J., Johns M., Kampfer T. et al. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection // Circ. Res. — 2012. — Vol. 110, № 1. — P. 20-33.
60. Jiang Y., Wang M., Huang K. et al. Oxidized low-density lipoprotein induces secretion of interleukin-1βby macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation // Biochem. Biophys. Res. Commun. — 2012. — Vol. 425, № 2. — P. 121-126.
61. Rajamaki K., Lappalainen J., Oorni K. et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation // PLoS ONE. — 2010. — Vol. 5, № 7. — Art. e11765.
62. Huang Z., Li W., Wang R. et al. 7-Ketocholesteryl-9-carboxynonanoate induced nuclear factor-kappa B activation in J774A.1 macrophages // Life Sci. — 2010. — Vol. 87, № 19–22. — P. 651-657.
63. Dasu M.R., Jialal I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors // Am. J. Physiol. Endocrinol. Metab. — 2011. — Vol. 300, № 1. — P. E145-E154.
64. Ishiyama J., Taguchi R., Yamamoto A., Murakami K. Palmitic acid enhances lectin-like oxidized LDL receptor (LOX-1) expression and promotes uptake of oxidized LDL in macrophage cells // Atherosclerosis. — 2010. — Vol. 209, № 1. — P. 118-124.
65. Sanson M., Distel E., Fisher E.A. HDL induces the expression of the M2 macrophage markers arginase 1 and fizz-1 in a STAT6-dependent process // PLoS ONE. — 2013. — Vol. 8, № 8. — Art. e74676.
66. De Paoli F., Staels B., Chinetti-Gbaguidi G. Macrophage phenotypes and their modulation in atherosclerosis // Circ. J. — 2014. — Vol. 78, № 8. — P. 1775-1781.
67. Kappus M.S., Murphy A.J., Abramowicz S. et al. Activation of liver X receptor decreases atherosclerosis in Ldlr mice in the absence of ATP-binding cassette transporters A1 and G1 in myeloid cells // Arterioscler. Thromb. Vasc. Biol. — 2014. — Vol. 34, № 2. — P. 279-284.
68. Liao X., Sluimer J.C., Wang Y. et al. Macrophage autophagy plays a protective role in advanced atherosclerosis // Cell. Metab. — 2012. — Vol. 15, № 4. — P. 545-553.
69. Shah M.S., Brownlee M. Molecular and cellular mecha–nisms of cardiovascular disorders in diabetes // Circ. Res. — 2016. — Vol. 118, № 11. — P. 1808-1829.
70. Fadini G.P., Menegazzo L., Scattolini V. et al. A perspective on NETosis in diabetes and cardiometabolic disorders // Nutr. Metab. Cardiovasc. Dis. — 2016. — Vol. 26. — P. 1-8.
71. Wong S.L., Demers M., Martinod K. et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healin // Nat. Med. — 2015. — Vol. 21. — P. 815-819.
72. Lee H.M., Kim J.J., Kim H.J. et al. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes // Diabetes. — 2013. — Vol. 62. — P. 194-204.
73. Guo H., Callaway J.B., Ting J.P. Inflammasomes: mecha–nism of action, role in disease, and therapeutics // Nat. Med. — 2015. — Vol. 21. — P. 677-687.
74. Misawa T., Takahama M., Kozaki T. et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome // Nat. Immunol. — 2013. — Vol. 14. — P. 454-460.
75. Iyer S.S., He Q., Janczy J.R. et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation // Immunity. — 2013. — Vol. 39. — P. 311-323.
76. Im S.S., Yousef L., Blaschitz C. et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a // Cell Metab. — 2011. — Vol. 13. — P. 540-549.
77. Du X., Edelstein D., Obici S. et al. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation // J. Clin. Invest. — 2006. — Vol. 116. — P. 1071-1080.
78. Ait-Oufella H., Taleb S., Mallat Z., Tedgui A. Recent advances on the role of cytokines in atherosclerosis // Arterioscler. Thromb. Vasc. Biol. — 2011. — Vol. 31. — P. 969-979.
79. Kimura T., Tse K., Sette A., Ley K. Vaccination to modulate atherosclerosis // Autoimmunity. — 2015. — P. 1-9.
80. Wolf D., Zirlik A., Ley K. Beyond vascular inflammation — recent advances in understanding atherosclerosis // Cell. Mol. Life Sci. — 2015. — Vol. 72, № 20. — P. 3853-3869.

/37-1.jpg)