
Журнал «Боль. Суставы. Позвоночник» Том 9, №2, 2019
Вернуться к номеру
Остеопетроз: класифікація, патоморфологія, генетичні порушення, клінічні прояви (огляд літератури та власне клінічне спостереження)
Авторы: Поворознюк В.В., Дєдух Н.В., Бистрицька М.А., Мусієнко А.С.
Державна установа «Інститут геронтології імені Д.Ф. Чеботарьова НАМН України», м. Київ, Україна
Рубрики: Ревматология, Травматология и ортопедия
Разделы: Справочник специалиста
Версия для печати
Остеопетроз — спадкове захворювання з автосомно-рецесивним чи автосомно-домінантним типом успадкування, спричинене порушенням функціональної активності остеокластів внаслідок мутації генів. У статті на основі аналізу літературних джерел систематизовані дані про етіологію, класифікацію, патоморфологію, генні порушення і висвітлені сучасні підходи до лікування остеопетрозу. Описано три типи остеопетрозу з різним ступенем вираженості порушень у скелеті та тяжкості патології. Подані основні патоморфологічні зміни у структурній організації кісткової тканини, відзначені особливості стану остеокластів залежно від мутації генів, які контролюють їх функціональну активність. Протоколів лікування цієї патології немає, але проводиться розробка методів лікування на основі використання гемопоетичних стовбурових клітин. Наведено клінічний приклад пацієнтки з остеопетрозом.
Остеопетроз — наследственное заболевание с аутосомно-рецессивным или аутосомно-доминантным типом наследования, вызванное нарушением функциональной активности остеокластов за счет мутации генов. В статье на основе анализа литературных источников систематизированы данные об этиологии, классификации, патоморфологии, генных нарушениях и освещены современные подходы к лечению остеопетроза. Описаны три типа остеопетроза с различной степенью выраженности нарушений в скелете и тяжестью патологии. Представлены основные патоморфологические изменения в структурной организации костной ткани, отмечены особенности состояния остеокластов в зависимости от мутации генов, контролирующих их функциональную активность. Протоколов для лечения этой патологии не имеется, но ведется разработка методов лечения на основе использования гемопоэтических стволовых клеток. Представлен клинический пример пациентки с остеопетрозом.
Osteopetrosis is a hereditary disease with an autosomal recessive or autosomal dominant type of inheritance, caused by a disruption in the functional activity of osteoclasts due to gene mutation. The article systematizes data on etiology, classification, pathomorphology, gene disorders based on the analysis of 38 sources of literature, and deals with the modern approaches to the treatment of osteopetrosis. Three types of osteopetrosis with different severity degrees of skeletal disorders and pathological severity are described. The main pathomorphological changes in the structural organization of bone tissue are presented and features of the state of osteoclasts are shown depending on the mutation of genes controlling their functional activity. There are no protocols for the treatment of this pathology, but treatment methods based on the use of hematopoietic stem cells are under development. The paper presents with clinical case report of a patient with marble bone disease.
остеопетроз; класифікація; патоморфологія; остеобласти; генні порушення; діагностика; лікування
остеопетроз; классификация; патоморфология; остеокласты; генные нарушения; диагностика; лечение
osteopetrosis; classification; pathomorphology; osteoclasts; gene disorders; diagnosis; treatment
Вступ
Уперше остеопетроз описав Альберс-Шенберг (Albers-Schonberg) 1904 року [1]. Остеопетроз (вроджений сімейний остеосклероз, «мармурова» хвороба, хвороба Альберс-Шенберга) — це загальна назва цілої низки рідкісних генних захворювань, які характеризуються склерозом скелета [2–7]. Дотепер відомо як мінімум одинадцять форм захворювання з різними типами успадкування та тяжкості. Захворюваність на автосомно-рецесивний остеопетроз становить 1 : 250 000 новонароджених, а на автосомно-домінантний остеопетроз — 1 : 20 000 новонароджених [6].
Мета огляду: систематизувати дані щодо етіології, класифікації, патоморфології генних порушень та підходів до лікування у хворих на остеопетроз, подати власне клінічне спостереження.
Інформаційний пошук було проведено у пошукових базах Google, GoogleScolar, AcademicResoursIndex, PubMed, РИНЦ із використанням ключових слів: остеопетроз, остеокласти, етіологія, класифікація, генні порушення, лікування.
Етіологія остеопетрозу
Етіологія та патогенез остеопетрозу потребують подальшого вивчення. Як основний патогенетичний механізм цієї хвороби розглядають порушення розвитку та функції остеокластів.
Остеокласти — це високоспеціалізовані клітини, які резорбують кістковий мінеральний та органічний матрикс. Ці процеси мають вирішальне значення для реконструкції кісток, підтримки їх біомеханічної міцності та мінерального гомеостазу. Скелет дорослої людини повністю оновлюється кожні 10 років [8].
Остеокласти походять від мононуклеарних клітин-попередників із мієлоїдної лінії гематопоезу, з яких також формуються макрофаги та моноцити. Термін життя остеокластів нетривалий і становить близько 14 діб.
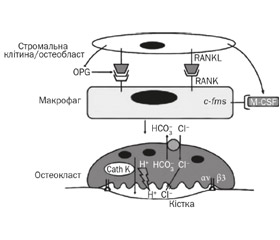
Для диференціації остеокластів необхідні рецептор-активатор ядерного фактора-kВ ліганд (RANKL) та макрофагальний колонієстимулюючий фактор (M-CSF), які продукуються клітинами остеобластичного диферону. Активація клітин-попередників та остеокластів відбувається шляхом взаємодії RANKL із рецепторами RANK остеобластів. Остеопротегерин — білок, синтезований клітинами остеобластичної лінії, є розчинним рецептором, який зв’язує RANKL, запобігаючи його взаємодії з RANK остеобластів, а отже й активації остеокластів та резорбції кістки.
Для виконання функції резорбції остеокласти повинні прикріпитися до поверхні кісткової тканини гофрованою кайомкою за рахунок унікально організованого цитоскелета та гетеродимера avb3 інтегрину (рецептор вітронектину). Компоненти, що сприяють утворенню avb3-асоційованого комплексу, складаються з молекул сигналізації — c-Src, Syk, Dap12, Slp76, Vav3 та Rac. Втрата будь-якої з цих молекул сигналізації призводить до порушення формування молекули інтегрину avb3 та дисфункції остеокласта. Після прикріплення остеокласта за рахунок інтегрину до поверхні кістки з відростків цитоплазми формується актинове кільце, що відокремлює мікросередовище від загального позаклітинного простору, тобто утворюється резорбційна лакуна. Формування актинового кільця є відмінною рисою цитоскелетної організації остеокласта.
Лакуни резорбції формуються внаслідок виділення ферментів, які руйнують органічний і мінеральний матрикси (рис. 1). Внутрішньоклітинний рН остеокласта підтримується за допомогою карбоангідрази електронейтральним обміном іонів НСО3–/Сl–. У руйнуванні мінерального матриксу бере участь соляна кислота, яка утворюється в лакунах резорбції з іонів хлору, що надходять через білки хлор-специфічного іонного каналу (CLCN7) на мембрані остеокластів, а також через остеокласт-специфічну АТФ протонну помпу [9]. Органічний матрикс руйнується катепсином K. Мутації генів, які відповідають за диференціацію остеокластів та їхню функціональну активність, призводять до остеопетрозу.
/136-1.jpg)
Хвороба розвивається внаслідок зменшення кількості остеокластів або часткової чи повної відсутності їх функції, що призводить до порушення резорбції кісток. У двох третин хворих остеокласти формуються, однак вони не здатні ефективно резорбувати кістку. Формування остеокластів може бути також порушене через нездатність до диференціації клітин-попередників. Порушення функції остеокластів обумовлене мутацією як мінімум у 10 генах, які були ідентифіковані як каузальні для остеопетрозу та виявлені в 70 % пацієнтів з остеопетрозом [11].
Найбільш досліджені мутації в генах CLCN7, CAІІ (карбоангідрази ІІ), TCIRG1 (субодиниця V-ATФази помпи) та OSTM1 (остеопетроз, асоційований із трансмембранним протеїном). Мутації порушують регуляторні механізми рН органели і секрецію кислот, що впливає на здатність остеокластів до резорбції кістки [6].
На рис. 2 наведені дані, які відображають складну регуляцію диференціації та функціональної активності остеокластів за рахунок внутрішньоклітинних сигнальних молекул, рецепторів RANKL — RANK, цитокінів та ферментів. Мутації генів, що забезпечують функцію цієї клітини, — шлях до розвитку остеопетрозу.
/137-1.jpg)
Захворювання може успадковуватися за різними типами: автосомно-рецесивним та автосомно-домі–нантним.
Патоморфологія кістки
При дослідженні кісток у хворих на остеопетроз виявляють значні осередки кісткоутворення як у ділянках кістково-мозкового каналу, так і в зовнішній частині трубчастих кісток, а також у міжтрабекулярних просторах трабекулярної кістки. Структурна організація новоутвореної кісткової тканини неоднорідна, мозаїчна, з вираженими патологічними змінами. Пластинчаста кісткова тканина з порушеними лініями цементації перемежовується з грубоволокнистою кістковою тканиною. У кістковому мозку розташовуються кісткові та хрящові острівці або скупчення остеоїду [3]. Кісткові трабекули трабекулярної кістки потовщені, що призводить до звуження міжтрабекулярних просторів [12]. Щільність остеокластів у кістковому мозку при різних типах остеопетрозу може відрізнятися — від низької до нормальної, однак осередки резорбції кістки поодинокі, що відображає порушення резорбційної функції остеокластів.
На основі проведення кісткової біопсії та генетичного аналізу у хворих на остеопетроз було виявлено, що значна кількість остеокластів (нормальний рівень або навіть підвищений) у кістковому мозку може свідчити про порушення їх функції, здебільшого внаслідок мутації генів TCIRG1, CLCN7, SNX10 та OSTM1, а наявність поодиноких остеокластів або їх відсутність у кістковому мозку пов’язані з рідкісним типом остеопетрозу, викликаним мутаціями генів RANK та RANKL [3].
Класифікація остеопетрозу
Для практичного клінічного застосування різні форми остеопетрозу можуть бути класифіковані відповідно до їхніх клінічних характеристик, тяжкості клінічних проявів, гістології кісткового мозку та генетичної основи [3, 13].
Найбільш тяжким є автосомно-рецесивний тип успадкування, який може бути пов’язаний із дефектами гена a-субодиниці АТФази — TCIRG1 (класичний тип) і гена остеопетрозасоційованого протеїну — OSTM1 (нейропатичний тип), карбоангідрази ІІ типу — KA-ІІ (з нирковим тубулярним ацидозом) [3]. Автосомно-рецесивний остеопетроз також спричиняється мутаціями у CLCN7 і рідше у SNX10, RANK і RANKL. Встановлено, що порушення сигнального шляху RANK-RANKL-OPG, орієнтованого на диференціацію й активацію остеокластів, призводять до автосомно-рецесивної форми остеопетрозу внаслідок скорочення кількості та функціональних можливостей остеокластів [14]. При проведенні біопсії кісткового мозку відзначається зниження кількості клітин, що свідчить про редукцію гемопоезу.
Автосомно-рецесивний остеопетроз може проявлятися вже на ембріональному етапі, а у новонароджених та грудних дітей простежують прогресуючу анемію, гепатоспленомегалію, макроцефалію або гідроцефалію, а також стиснення нервів, що супроводжується сліпотою та глухотою. Цю форму визначають як злоякісний остеопетроз. При неонатальній формі остеопетрозу виживання низьке. Тривалість життя становить не більше 2–10 років. Захворювання характеризується остеосклерозом із множинними переломами, може призвести до остеомієліту, втрати зору та порушення гемопоезу. Характерним є розширення лицьового черепа, порушення анатомії носа та губ. Психічна затримка та прогресуюча нейродегенерація можуть виникати внаслідок мутації генів CLCN7 та OSTM1 [9, 15]. У гені CLCN7 виявлено 20 мутацій. Для цього типу остеопетрозу є характерним розвиток кісткових дисплазій.
Проміжна форма остеопетрозу клінічно та генетично неоднорідна, характеризується менш вираженим, але також тяжким перебігом, із домінантним або рецесивним успадкуванням. Зниження активності гена, що кодує білок CLCN7, внаслідок мутації призводить до розвитку декількох форм рецесивного остеопетрозу та автосомно-домінантного остеопетрозу типу II [16, 17]. Із дефектом гена CLCN7, гена білкового домена, гомологічного плексетрину 1 (PLEKHM 1) також пов’язують проміжний тип остеопетрозу [6]. Проміжна рецесивна форма проявляється кальцифікацією кісткового мозку, нирковим тубулярним ацидозом, що зумовлене мутацією гена карбонангідрази ІІ (KA II). У цих пацієнтів часто спостерігаються психічні розлади [18]. Також остеопетроз може бути викликаний мутаціями генів RANK, SNX10 та TCIRG1 [6, 9, 19–21]. Інші проміжні форми характеризуються помірним остеосклерозом, низьким зростом та низькоенергетичними переломами.
Помірний автосомно-домінантний тип остеопетрозу характеризується неоднорідними та пізніми проявами, визначається як доброякісна доросла форма. Легкі форми остеопетрозу приблизно в половини хворих можуть мати безсимптомний перебіг [13], а в інших випадках виникають патологічні переломи навіть при незначних травмах, які нерідко бувають єдиним проявом захворювання в дорослих. Зрощення переломів уповільнене. Захворювання супроводжується болем у кістках, ускладнюється остеомієлітом і ураженням черепно-мозкових нервів [2, 22]. Ламкість кісток підвищена внаслідок порушення архітектоніки й атипового кісткоутворення. Найчастішим ускладненням остеопетрозу є атрофія зорового нерва внаслідок розростання кісткової тканини в ділянках отворів і каналів, що призводить до сліпоти. Автосомно-домінантний тип захворювання частіше зустрічається в дітей старшого віку та дорослих, основні його прояви — патологічні переломи. При пізній формі остеопетрозу ураження кісток зазвичай обмежене [23]. Рання смерть із цим типом остеопетрозу зустрічається рідко, тривалість життя в більшості випадків звичайна, однак якість життя порушена [10].
Розрізняють два типи дорослої форми остеопетрозу. У разі автосомно-домінантного остеопетрозу І типу фенотипові прояви патології кісткової тканини, викликані місенс-мутацією (точкова мутація, в результаті якої змінений кодон починає кодувати іншу амінокислоту) в LRP5 [24–26]. Автосомно-домінантний остеопетроз II типу може бути викликаний місенс-мутацією CLCN7 [27].
Типи остеопетрозу генетично детерміновані і можуть бути представлені як остеокласт-автономний остеопетроз або остеокласт-неавтономний остеопетроз [28, 29]. При неавтономному типі генетичний дефект може бути пов’язаний із клітинами, що впливають на диференціювання клітин — попередників остеокластів або на функцію зрілих клітин. При остеокласт-автономному остеопетрозі молекулярний дефект наявний або в самих остеокластах, або в клітинах — попередниках остеокластів.
Хоча практично всі форми остеопетрозу генетично детерміновані, хвороба може бути індукована в дітей, яких лікують бісфосфонатами, що сприяють апоптозу остеокластів [30, 31]. У разі остеопетрозу формуються кісткові дисплазії.
Діагностика
Діагностика остеопетрозу включає оцінку клінічних та рентгенологічних даних із використанням кісткової денситометрії. Для підтвердження діагнозу важливу роль відіграє генетичне дослідження.
Діагноз остеопетрозу ґрунтується на характерних рентгенологічних ознаках, детально описаних Z. Stark та R. Savarirayan [6]:
— наявність дифузного остеосклерозу в кістках черепа, хребта, таза і кінцівок;
— метафізи довгих трубчастих кісток потовщені (колби Ерленмейєра), ущільнені, з лінійними дефектами;
— феномен «кістка в кістці» — подвійний контур кісток, особливо в хребцях та фалангах пальців;
— вогнищевий остеосклероз кісток склепіння черепа, таза і замикальних пластин хребців — хребці типу «сендвіч», «смугастий» хребет.
На підставі рентгенологічних даних виділяють два типи автосомно-домінантного остеопетрозу [2]. Для автосомно-домінантного остеопетрозу I типу характерне потовщення склепіння черепа, а при II типі переважні ознаки — «смугастий» хребет та віялоподібні смугастості в крилах клубових кісток, а також ущільнення губчастої речовини кісток. При обох типах остеопетрозу звужуються кістковомозкові порожнини. При захворюванні уражаються кістки всього скелета, але особливо виражені порушення виявляються в довгих кістках, кістках черепа, хребта, ребер і таза.
У разі обстеження пацієнтів на двофотонній рентгенівській абсорбціометрії показники мінеральної щільності кісткової тканини (МЩКТ) у 2–4 рази і більше перевищують нормативні показники.
У гематологічному аналізі проводять підрахунок клітин крові, включаючи кількість ретикулоцитів у мазку крові, а також визначають лактатдегідрогеназу в сироватці крові, що є обов’язковим для оцінки ступеня гематологічного порушення. Виявлення зниження показників гемоглобіну, ретикулоцитів та тромбоцитів співвідносять з порушеннями функціонування кісткового мозку. На відміну від цього збільшення кількості лейкоцитів, а також поява незрілих гранулоцитів, підвищений рівень лактатдегідрогенази зазвичай вказують на екстрамедулярний гемопоез.
При лабораторній діагностиці в дорослих пацієнтів рівень кальцію (загального та іонізованого), фосфору, активність кислої фосфатази в плазмі крові зазвичай у межах норми. Однак ці показники необхідно аналізувати для виявлення гіперкальціємії. У дітей частіше спостерігається гіпофосфатемія і помірна гіпокальціємія. Активність кислої фосфатази в плазмі зазвичай підвищена.
Лікування
Протоколів щодо лікування остеопетрозу не існує. Лікування цієї патології симптоматичне, використовується підтримуюча терапія. Часті переломи і вторинні ускладнення, як-от уповільнена консолідація уламків або незрощення перелому, вимагають спостереження в ортопеда й потребують особливого підходу до хірургічного лікування [5, 32].
Останніми роками при тяжких формах автосомно-рецесивного остеопетрозу застосування трансплантації гемопоетичних стовбурових клітин дало можливість досягти в 73 % випадків 5-річного виживання пацієнтів [23]. Цей вид терапії використовують з урахуванням гематологічного походження остеокластів. Переважно позитивні результати (> 50 %) отримані в разі лікування автосомно-рецесивного остеопетрозу, але відзначені деякі небажані ускладнення, а саме: порушення зору в ранньому періоді після трансплантації [8, 12, 33]. Аналогічні позитивні результати отримані в разі лікування пацієнтів із мутацією карбоангідрази ІІ [10]. Однак у разі мутації генів рецепторів RANK та RANKL цей метод не є ефективним.
Є поодинокі клінічні підтвердження, що високі дози кальцитріолу можуть знизити симптоматичні прояви остеопетрозу [23], а використання рекомбінантного паратгормону ефективне в разі переломів [34, 35], однак доказової бази щодо використання цих препаратів наразі немає. Останнім часом проводяться доклінічні випробування нових видів терапії, а саме замісної терапії з використанням RANKL та деносумабу в разі остеопетрозу з RANK-мутацією [6, 7, 36].
Безумовно, успіх розробки препаратів для лікування цього захворювання обмежений відсутністю тваринних моделей, які б за характером перебігу відтворювали захворювання людини. Лише 2017 року вперше було розроблено на мишах модель автосомно-домінантного остеопетрозу ІІ типу, особливістю якої була зміна ступеня тяжкості фенотипу, що нагадує широкі варіації фенотипу, які спостерігаються в пацієнтів із цим захворюванням [37]. Автори прогнозують, що ця модель допоможе ідентифікувати мутантні гени або фактори, які впливають на тяжкість і пенетрантність цього типу остеопетрозу, та буде сприяти випробуванню інноваційних методів лікування цієї тяжкої хвороби.
Клінічний випадок
Пацієнтка Р., 1952 р. н. Скарги на інтенсивний біль у попереково-крижовому відділі хребта, колінних та гомілковостопних суглобах, який посилюється при ходьбі.
Анамнез захворювання
Вважає себе хворою з 1972 року, коли під час проведення флюорографії було зареєстроване значне ущільнення кісткової тканини й установлений діагноз «мармурова хвороба». На момент встановлення діагнозу жодних скарг у пацієнтки не було. Перші скарги — біль у попереку та головний біль — стали турбувати 1979 року (27 років) після других пологів. Пацієнтка спостерігалась у ДУ «Інститут ортопедії і травматології», регулярно отримувала курси симптоматичного лікування. 2016–2017 рр. значно збільшилась інтенсивність больового синдрому в поперековому відділі хребта та суглобах нижніх кінцівок.
Анамнез життя
Жінка 65 років, не палить і не палила, алкоголь не вживає. Професійні шкідливі чинники заперечує. Алергологічний анамнез не обтяжений. Менархе з 14 років, менопауза у віці 49 років, замісну гормональну терапію не вживала. Інші супутні захворювання та прийом будь-яких лікарських засобів заперечує.
Об’єктивний статус
Гіперстенічної статури, зріст — 164 см, маса тіла — 96 кг, ІМТ — 35,7 кг/м2. Шкіра, видимі слизові фізіологічного кольору. Периферичні лімфатичні вузли не збільшені. Пульс — 68 уд/хв, АТ — 130/80 мм рт.ст., ЧД — 14 за хв. Тони серця ритмічні, приглушені, акцент ІІ тону на аорті. У легенях дихання везикулярне, хрипів немає. Живіт при пальпації м’який, безболісний. Нижня межа печінки — біля краю реберної дуги. Селезінка не пальпується. Периферичних набряків немає. Діурез достатній, випорожнення в нормі.
Хода уповільнена, без додаткових засобів опори. Постава звичайна, фізіологічні вигини хребта згладжені. Рухи в шийному та поперековому відділах хребта помірно обмежені, помірно болючі, супроводжуються хрустом. Паравертебральні м’язи гіпертонічні, паравертебральні точки болючі на рівні С4–6, L4–S1. Рухи у суглобах верхніх кінцівок збережені в повному обсязі, болючі. Колінні суглоби набряклі, без ознак запалення. Рухи в колінних суглобах болючі, у повному обсязі, супроводжуються хрустом. Пальпаторно: болючість проекції суглобової щілини колінних суглобів. Суглоби кистей помірно набряклі. Рухи в них болючі. Сила та тонус м’язів кінцівок збережені.
Результати лабораторних досліджень
Результати загальноклінічних досліджень — без клінічно значущих відхилень. Результати загального аналізу крові: еритроцити — 4,76 (норма: 3,8–5,8 • 1012/л), гемоглобін — 140 (120–140 г/л), лейкоцити — 5,4 (4–10 • 109/л), лімфоцити — 37,9 (17,0–48,0 %), моноцити — 5,2 (4,0–10,0 %), гранулоцити — 56,9 (43,0–76,0 %), тромбоцити — 272 (150–400 • 109), швидкість осідання еритроцитів — 28 (2–18 мм/год). Біохімічний аналіз крові: альбумін — 40,6 (32,0–52,0 г/л), аланінамінотрансфераза — 27 (до 41 ммоль/л), аспартатамінотрансфераза — 24 (до 40 ммоль/л), глюкоза крові — 5,9 (3,8–6,1 ммоль/л), сечовина — 5,0 (2,5–8,3 ммоль/л), креатинін — 70,4 (53,0–97,0 мкмоль/л). Показники метаболізму кісткової тканини без відхилень: Ca загальний — 2,36 (2,15–2,58 ммоль/л), вітамін D загальний (25(ОН)D) — 34,41 (30,0–50,0 нг/мл).
Рівні маркерів ремоделювання кісткової тканини підвищені: пропептиди проколагену I типу (P1NP) — 157,4 (16,3–73,9 нг/мл), b-термінальні телепептиди колагену I типу (b-СТх) — 1,13 (< 1,008 нг/мл).
Результати інструментальних досліджень
ЕКГ (10.07.2017) — ритм синусний, правильний, ЧСС 71 уд/хв. Електрична вісь серця — горизонтально. Ознаки гіпертрофії лівого шлуночка, лівого передсердя.
УЗД органів черевної порожнини (18.07.2017) — жирова дистрофія печінки, ознаки хронічного холециститу, дифузні зміни в підшлунковій залозі.
Рентгенографія шийного відділу хребта (12.07.2017) — остеопетроз, остеохондроз дисків С4–5, С5–6, спондильоз.
Двофотонна рентгенівська абсорбціометрія (13.07.2017) — значне збільшення МЩКТ на рівні всіх обстежених ділянок: МЩКТ поперекового відділу хребта за показником Т = 11,0 SD, МЩКТ шийки стегнової кістки за показником Т = 8,4 SD, МЩКТ усього скелета за показником Т = 13,8 SD та МЩКТ кісток передпліччя за показником Т = 5,9 SD (рис. 3). Нормальні значення Т-критерію в межах від –1 до +1. Показник якості трабекулярної кісткової тканини (TBS) — 1,630.
/139-1.jpg)
Діагноз «Остеопетроз (мармурова хвороба) зі статико-динамічними порушеннями та вираженим іритативно-больовим синдромом» був встановлений на підставі клінічних та рентгенологічних ознак без генетичного тестування.
Таким чином, остеопетроз — це спадкове захворювання, в основі якого лежить остеосклероз. Патологія зумовлена порушенням диференціації та активності остеокластів, що проявляється змінами структурної організації кістки, її компактизацією і підвищеною крихкістю, з підвищеним ризиком переломів. Остеопетроз — генетично обумовлене захворювання з автосомно-рецесивним та автосомно-домінантним типом успадкування, з більш тяжкими проявами в разі автосомно-рецесивного типу.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів при підготовці даної статті.