
Международный эндокринологический журнал 3 (67) 2015
Вернуться к номеру
Взаимное влияние тиреоидного и углеводного обмена. Парадигмы и парадоксы
Авторы: Бобрик М.И. — Национальный медицинский университет им. А.А. Богомольца, г. Киев
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
Статья посвящена влиянию гормонов щитовидной железы на углеводный обмен. Влияние тиреоидных гормонов на углеводный обмен зависит от преморбидного статуса, наследуемой предрасположенности к инсулинорезистентности и других факторов. Многим пациентам с сахарным диабетом приходится корректировать потребность в инсулине при развитии тиреотоксикоза или гипотиреоза. Своевременная коррекция нарушений функции щитовидной железы приводит к восстановлению метаболических изменений.
Стаття присвячена впливу гормонів щитоподібної залози на вуглеводний обмін. Вплив тиреоїдних гормонів на вуглеводний обмін залежить від преморбідного статусу, успадкованої схильності до інсулінорезистентності та інших факторів. Багатьом пацієнтам із цукровим діабетом доводиться коректувати потребу в інсуліні при розвитку тиреотоксикозу або гіпотиреозу. Своєчасна корекція порушень функції щитоподібної залози призводить до відновлення метаболічних змін.
The article considers the influence of thyroid hormones on carbohydrate metabolism. The impact of thyroid hormones on carbohydrate metabolism depends on premorbid status, inherited predisposition to insulin resistance and other factors. Many patients with diabetes mellitus have to adjust the need for insulin in the development of hyperthyroidism or hypothyroidism. Timely correction of the thyroid dysfunctions lead to the restoration of metabolic changes.
тиреоидные гормоны, щитовидная железа, углеводный обмен, сахарный диабет, инсулинорезистентность.
тиреоїдні гормони, щитоподібна залоза, вуглеводний обмін, цукровий діабет, інсулінорезистентність.
thyroid hormones, thyroid gland, carbohydrate metabolism, diabetes mellitus, insulin resistance.
Статья опубликована на с. 127-132
Общепризнанным является утверждение, что распространенность сахарного диабета (СД) и тиреоидной дисфункции в мире растет. Свой вклад в видоизменение морбидной структуры вносит факт наличия в Украине йодного дефицита.
По данным Международной диабетической федерации, распространенность СД в мире составляет 382 млн человек, и по самым оптимистичным прогнозам она составит около полумиллиарда человек к 2035 году. Неинфекционная эпидемия СД набирает обороты. Прирост заболевших с 2009 по 2013 год (за 4 года) составил 98 млн чел., тогда как с 1990 по 2000 год (за 10 лет) такой прирост был 40 млн чел. [1].
Определены закономерности эндокринной патологии в регионах легкого йодного дефицита. В йододефицитном регионе по мере старения у человека повышается вероятность формирования многоузлового зоба с избыточным функционированием щитовидной железы (ЩЖ). Частота гипотиреоза и СД также будет возрастать по мере старения человека. В регионе с нормальным йодным обеспечением с годами нарастает частота гипотиреоза, но не увеличивается частота тиреотоксикоза. Поэтому в Украине, регионе с йодным дефицитом, ожидаем по мере старения роста числа больных с СД, тиреотоксикозом на фоне многоузлового зоба и гипотиреоза.
Распространенность тиреопатий в йододефицитном регионе приводится в разных источниках в интервалах от 9 до 16 % (частота гипотиреоза в исходе аутоиммунного тиреоидита), 15 % лиц имеют в молодом возрасте антитела к тиреопероксидазе (АТ-ТПО). Факт проживания в регионе с йодным дефицитом увеличивает частоту людей с АТ-ТПО и антителами к тиреоглобулину (АТ-ТГ). Частота болезни Грейвса — Базедова составляет 0,1 %. Таким образом, около 24 % людей имеют признаки аутоиммунной тиреопатии [6].
Влияние тиреоидных гормонов на углеводный обмен
Начиная с 50-х годов прошлого века публиковалось много исследований, посвященных связи заболеваний ЩЖ и СД. К 80-м годам было сформулировано понятие «инсулинорезистентность». Различные исследователи в работах как по гипотиреозу, так и по тиреотоксикозу говорили о наличии сопутствующей инсулинорезистентности. При этом любой клиницист понимал, что тиреотоксикоз с большой степенью вероятности ухудшит параметры углеводного обмена, а гипотиреоз может создать проблему гипогликемии. Оба эти феномена объяснимы, поскольку тиреоидные гормоны не только антагонисты инсулина, как мы привыкли думать, но, кроме того, по влиянию на периферические ткани они являются синергистами инсулина и способствуют транспорту и утилизации глюкозы.
Помимо непосредственного влияния на печень и мышцы, происходит активация симпатической нервной системы (НС) через гипоталамические центры, влияние на расход энергии и аппетит, на бурую жировую ткань у взрослого человека.
Сочетание таких метаболических нарушений, как ожирение, дислипидемия, артериальная гипертензия и инсулинорезистентность, обозначается термином «метаболический синдром», который сопровождается увеличением риска сердечно-сосудистых заболеваний [3]. Однако эти же компоненты метаболического синдрома встречаются и при явном и субклиническом гипотиреозе [2, 4, 5]. Это отчасти подтверждает предположение о том, что снижение функции ЩЖ, даже когда тиреоидные гормоны находятся в рамках нормальных значений, может увеличивать сердечно-сосудистый риск, связанный с метаболическим синдромом.
Гормоны щитовидной железы и углеводный обмен
Прежде чем перейти к обсуждению общих патофизиологических механизмов СД и нарушений функции ЩЖ, необходимо заметить, что гормоны ЩЖ оказывают выраженное влияние на регуляцию гомеостаза глюкозы. Это воздействие заключается в изменении уровня инсулина и гормонов-антагонистов в крови, абсорбции глюкозы в кишечнике, продукции глюкозы печенью и утилизации ее периферическими тканями (жировой и мышечной) [7].
Стимулируя не только глюконеогенез в печени, но и инсулинозависимый транспорт глюкозы в мышечную и жировую ткань, тиреоидные гормоны оказывают прямое влияние на транскрипцию генов в печени и непрямое влияние — через центральный симпатический путь, и таким образом усиливают продукцию глюкозы печенью. Влияние гормонов ЩЖ на углеводный и липидный обмен также осуществляется посредством 5'аденозин-монофосфат-активируемой протеинкиназы, контролирующей энергетический баланс клетки. В последнее время широко изучается не только модуляция чувствительности к инсулину, но и ответ тиреоидных гормонов на аппетит и потребление энергии [8].
Прямые эффекты тиреоидных гормонов на печень
Установлено, что тиреоидные гормоны оказывают влияние на некоторые гены гепатоцитов, участвующие в глюконеогенезе, метаболизме гликогена и передаче инсулинового сигнала. Примером такого влияния являются ферменты глюконеогенеза — пируваткарбоксилаза и фосфоенолпируваткарбоксикиназа. В митохондриях пируват под влиянием пируваткарбоксилазы карбоксилируется с образованием оксалоацетата [9]. Затем оксалоацетат превращается в фосфоенолпируват в ходе реакций декарбоксилирования и фосфорилирования, катализируемых фосфоенолпируваткарбоксикиназой [10], являющейся мишенью для трийод-тиронина (Т3). Более того, Т3 вызывает усиление экспрессии мРНК глюкозо-6-фосфатазы, конечного фермента глюконеогенеза и гликогенолиза, катализирующего гидролиз глюкозо-6-фосфата с образованием глюкозы [11].
Имеются данные, что тиреоидные гормоны снижают экспрессию мРНК протеинкиназы В, серин/треониновой киназы — продукта гена Akt2 — ключевой молекулы, участвующей в передаче инсулинового сигнала [11]. Akt2 участвует в синтезе гликогена в печени посредством ингибирования киназы-3 гликогенсинтазы, что приводит к активации гликогенсинтазы. Таким образом, снижение активности Akt2, в свою очередь приводящее к снижению синтеза гликогена, является примером влияния тиреоидных гормонов на печень в качестве антагонистов инсулина. Индукция мРНК бета-2-адренергических рецепторов и подавление РНК G-белка, ингибирующего аденилатциклазу под влиянием тиреоидных гормонов, приводит к потенцированию гликогенолитического и глюконеогенного эффектов адреналина и глюкагона [11].
Другим примером влияния тиреоидных гормонов на печень в качестве антагонистов инсулина является увеличение экспрессии транспортера глюкозы ГЛЮТ-2 в печени, что приводит к увеличению выхода глюкозы из печени в кровь [12].
Непрямые эффекты тиреоидных гормонов на печень
Недавно было описано действие Т3 на метаболизм глюкозы в печени через гипоталамус независимо от уровня в плазме гормонов, влияющих на углеводный обмен [13]. Показано, что селективное влияние Т3 на паравентрикулярное ядро гипоталамуса приводит к увеличению синтеза глюкозы и усилению выхода ее в кровь независимо от уровня Т3, инсулина и кортикостероидов в крови. Указанные эффекты реализуются через симпатические волокна, иннервирующие гепатоциты.
Прямые эффекты тиреоидных гормонов на периферические ткани
В периферических тканях тиреоидные гормоны регулируют экспрессию генов, влияющих на гликолиз и транспорт глюкозы. Но в отличие от влияния тиреоидных гормонов на печень в периферических тканях они оказывают действие, сходное с эффектами инсулина. В скелетных мышцах транспортер глюкозы ГЛЮТ-4 индуцируется тиреоидными гормонами, что приводит к увеличению базального и инсулиностимулированного транспорта глюкозы в мышцы. Кроме того, установлено, что в фибробластах кожи Т3 усиливает транскрипцию мРНК фактора 1, индуцируемого гипоксией (HIF-1), ключевого медиатора гликолиза. Другой точкой приложения тиреоидных гормонов является 1-a коактиватор гамма-рецептора, активируемого пролифераторами пероксисом (PGC-1a), основного транскрипционного регулятора митохондриального биогенеза, окисления жирных кислот и глюконеогенеза. Снижение экспрессии PGC-1a при снижении уровня тиреоидных гормонов может привести к увеличению внутриклеточного содержания липидов и ухудшению их окисления, что характерно для CД 2-го типа [14].
Эффекты Т3 зависят не только от его содержания в плазме, но и от его внутриклеточной концентрации в зависимости от активности дейодиназ. Так, снижение экспрессии и активности йодтиронин-дейодиназы 2-го типа (D2) ассоциировано с инсулинорезистентностью. В настоящее время проводятся исследования по изучению роли желчных кислот, мощных стимуляторов дейодиназы, и полиморфизма дейодиназы 2-го типа, приводящего к снижению активности этого фермента [2].
Тиреотоксикоз и инсулинорезистентность
При тиреотоксикозе увеличение базальной продукции глюкозы печенью и снижение печеночной чувствительности к инсулину компенсируется повышенной утилизацией глюкозы периферическими тканями. Обнаружено ускорение инсулиностимулированного окисления глюкозы в мышечной и жировой ткани. Тем не менее снижение инсулиностимулированной неоксидативной утилизации глюкозы в периферических тканях посредством подавления гликогеногенеза свидетельствует о том, что избыток гормонов ЩЖ может вызывать периферическую инсулинорезистентность, что было подтверждено в нескольких исследованиях. Одним из объяснений таких разночтений является тот факт, что при тиреотоксикозе утилизация глюкозы не увеличивается параллельно с увеличением кровотока. Другие авторы предполагают, что увеличение секреции биоактивных медиаторов (адипокинов) в жировой ткани, таких как интерлейкин-6 (ИЛ-6) и фактор некроза опухоли aльфа (ФНО-aльфа), объясняет развитие инсулинорезистентности при тиреотоксикозе [2, 8].
Что касается уровня инсулина в плазме, то при тиреотоксикозе определялся как нормальный или сниженный, так и повышенный уровень инсулина. Тиреотоксикоз сопровождается усиленной деградацией инсулина, а тяжелый тиреотоксикоз, посредством Т3-индуцированного апоптоза бета-клеток, может привести к необратимому повреждению инсулярного аппарата. Что касается уровня глюкагона, то его секреция и метаболический клиренс при тиреотоксикозе усиливаются. Это объясняет нормальный уровень глюкагона в плазме натощак у пациентов с тиреотоксикозом [8].
Также проводились исследования по оценке влияния субклинического тиреотоксикоза на чувствительность к инсулину. В одном из них не было выявлено разницы по чувствительности к инсулину между пациентами с раком ЩЖ, которые находились на заместительной или супрессивной терапии левотироксином. Тем не менее в других исследованиях как при эндогенном, так и при ятрогенном субклиническом тиреотоксикозе определялась инсулинорезистентность различной степени выраженности. Более того, у пациентов с эндогенным субклиническим тиреотоксикозом инсулинорезистентность оказалась более выраженной, чем в группе ятрогенного тиреотоксикоза, что объясняется продолжительностью тиреотоксикоза и более высоким уровнем Т3 по сравнению с терапией левотироксином [14].
Гипотиреоз и инсулинорезистентность
Механизмы возникновения инсулинорезистентности при гипотиреозе: нарушение транслокации ГЛЮТ-4, нарушение действия лептина на гипоталамус, снижение кровотока по отношению к экстракции глюкозы тканями, увеличение уровня циркулирующих свободных жирных кислот [13].
С другой стороны, выявлено, что при гипотиреозе подавление глюконеогенеза, приводящее к снижению продукции глюкозы печенью, компенсируется недостаточной утилизацией глюкозы мышцами и другими периферическими тканями.
У пациентов, прооперированных по поводу рака ЩЖ, на фоне прекращения приема левотироксина, в ходе инсулинотолерантного теста была выявлена инсулинорезистентость. В более ранних исследованиях при этом было продемонстрировано снижение утилизации глюкозы, выявленное с помощью эугликемического гиперинсулинемического клэмпа. Это же было выявлено у женщин с первичным гипотиреозом с использованием артериовенозной разницы по постпрандиальной глюкозе, полученной из лучевой артерии, вены подкожно-жировой клетчатки передней брюшной стенки и глубокой вены предплечья. Однако в других исследованиях связи между гипотиреозом и инсулинорезистентностью не было выявлено ни с использованием расчетного индекса инсулинорезистентности (HOMA-IR), показывающего чувствительность к инсулину натощак, ни при оценке кровотока в мышцах предплечья, ни при использовании комбинации последнего метода с эугликемическим гиперинсулинемическим клэмпом [14].
При гипотиреозе секреция инсулина может быть как в норме, так и незначительно снижена или повышена [5]. Последние данные свидетельствуют о том, что секреция инсулина при гипотиреозе снижена, так как после начала терапии левотироксином концентрации инсулина и проинсулина значительно увеличиваются. И напротив, при гипотиреозе было выявлено повышение глюкозоиндуцированной секреции инсулина с последующим ее снижением на фоне терапии левотироксином, поскольку при восстановлении эутиреоза снижается нагрузка на бета-клетки [15].
При субклиническом гипотиреозе в нескольких работах проводилась оценка индекса НОМА; инсулинорезистентность была выявлена в некоторых, но не во всех исследованиях. Тем не менее в исследованиях, в которых инсулинорезистентности при субклиническом гипотиреозе выявлено не было, авторы обнаружили гиперинсулинемию как первый признак нарушения метаболизма глюкозы. Было выявлено снижение инсулиностимулированного транспорта глюкозы в моноциты, что объясняется нарушением транслокации ГЛЮТ-4 на плазматической мембране.
Щитовидная железа и диабет
Сочетание СД 1-го типа с аутоиммунными заболеваниями (АИЗ) ЩЖ обозначается как аутоиммунный полигландулярный синдром; оба эти заболевания объединяет аутоиммунный генез. У пациентов с СД 1-го типа АТ-ТПО и АТ-ТГ встречаются чаще, чем в общей популяции. Повышенные уровни АТ-ТПО и тиреотропного гормона (ТТГ) являются предикторами нарушения функции ЩЖ у пациентов с СД. АТ-ТГ у пациентов с СД 1-го типа, образующиеся в ответ на эпитоп, единый для пациентов с АИЗ ЩЖ и СД 1-го типа, также являются предиктором тиреоидной дисфункции.
Выявлены гены, позволяющие объяснить связь между СД 1-го типа и АИЗ ЩЖ. К ним относятся прежде всего гены HLA, локализованные на коротком плече шестой хромосомы (6р21). Двукратное увеличение риска развития болезни Грейвса ассоциировано с гаплотипом HLA-DR3, в то время как риск развития СД 1-го типа, ассоциированный с этим же гаплотипом, увеличивается в 3–4 раза. Другим возможным геном, предрасполагающим к развитию АИЗ ЩЖ и СД 1-го типа, является ген PTPN22 и ген CTLA-4. Тирозиновая фосфатаза лимфоидных клеток, которая кодируется геном PTPN22, является негативным регулятором иммунного ответа, осуществляя дефосфорилирование белков, передающих сигнал от антигенных рецепторов Т-клеток (CD3) [16].
У трети взрослых пациентов с СД 1-го типа было выявлено нарушение функции ЩЖ; 20 % детей с СД 1-го типа имели положительные антитела к ЩЖ и 3–8 % — гипотиреоз. Более того, у 25 % беременных с СД 1-го типа развивается послеродовой тиреоидит.
Многие клинические рекомендации предлагают всем пациентам с СД 1-го типа проводить исследование уровня ТТГ и антител к ЩЖ с последующим ежегодным определением ТТГ, особенно у пациентов с АТ-ТПО, на предмет раннего выявления бессимптомных нарушений функции ЩЖ.
В отличие от СД 1-го типа при СД 2-го типа распространенность заболеваний ЩЖ, видимо, такая же, как и в общей популяции, поэтому необходимость скрининга на ТТГ в этой когорте пациентов до сих пор не определена. Тем не менее у пациентов с СД 2-го типа с положительными антителами к изоформе глутаматдекарбоксилазы (GAD) с молекулярной массой 65 кДа (GAD65), так называемым латентным аутоиммунным диабетом взрослых (LADA), повышен риск развития АИТ. У тучных детей с СД 2-го типа и аутоиммунным поражением бета-клеток (двойной диабет или диабет 1,5 типа) распространенность высокого уровня антител к ЩЖ и АИЗ ЩЖ сходна с таковой при СД 1-го типа. Вероятно, только высокий уровень антител к GAD ассоциирован с развитием АИЗ ЩЖ у пациентов с СД 1-го типа и LADA [16].
Установлено, что при развитии тиреотоксикоза у пациентов с СД 1-го типа потребность в инсулине увеличивается. Периферические ткани плохо справляются с гиперпродукцией глюкозы печенью, что приводит к нарушению толерантности к глюкозе (НТГ). Более того, было описано несколько случаев кетоацидоза. Гипотиреоз редко оказывает влияние на метаболический контроль у пациентов с СД. При гипотиреозе утилизация глюкозы снижена, однако вследствие сниженного выхода глюкозы из печени в кровь гипергликемия не развивается. Тем не менее иногда при гипотиреозе у пациентов с СД 1-го типа может снижаться потребность в инсулине и увеличиваться риск гипогликемий, и это может встречаться не только при явном, но и при субклиническом гипотиреозе. В одном из ретроспективных исследований было показано увеличение частоты гипогликемий у детей с СД 1-го типа и повышенным уровнем ТТГ [17].
Щитовидная железа и метаболический синдром
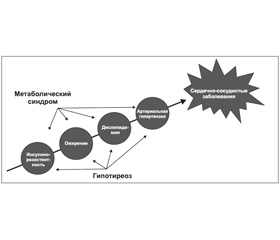
Метаболический синдром представляет собой совокупность факторов, определяющих сердечно-сосудистый риск, таких как инсулинорезистентность, ожирение, дислипидемия, повышение артериального давления (АД). Гипотиреоз также связан с перечисленными состояниями, которые могут развиваться и при нормальном уровне тиреоидных гормонов. В связи с этим можно предположить, что нарушение функции ЩЖ может усиливать риск сердечно-сосудистых заболеваний (ССЗ) у пациентов с метаболическим синдромом (рис. 1).
/131/131.jpg)
В литературе есть данные о более частом развитии субклинического гипотиреоза у лиц с метаболическим синдромом. Что касается распространения метаболического синдрома у пациентов с явным гипотиреозом, то по результатам исследования, проведенного в Мексике, стране с высокой распространенностью метаболического синдрома, в группе лиц без нарушения функции ЩЖ и лиц с субклиническим гипотиреозом распространенность метаболического синдрома оказалась одинаковой. Тем не менее в этом исследовании была установлена положительная корреляция уровня ТТГ с общим холестерином, триглицеридами и окружностью талии, тогда как уровень свободного тироксина (Т4) положительно коррелировал с липопротеинами высокой плотности и отрицательно коррелировал с окружностью талии, уровнем инсулина и HOMA-IR. В другом одномоментном исследовании приняли участие 100 пациентов с субклиническим гипотиреозом, 100 — с явным гипотиреозом и 200 лиц — без нарушения функции ЩЖ. Было показано, что метаболический синдром встречается чаще только в группе пациентов с клиническим гипотиреозом по сравнению с другими группами. Тем не менее количество пациентов с абдоминальным ожирением было также выше в группе субклинического гипотиреоза, чем в группе контроля. Возможным объяснением противоречивых результатов исследований является использование различных уровней ТТГ, на основании которых диагностируется субклинический гипотиреоз, различных критериев метаболического синдрома, обеспечения йодом, раса, возраст, пол и другие отличия изучаемых групп [14].
Как упоминалось ранее, существует связь между высоконормальным уровнем ТТГ и метаболическим синдромом. В одномоментном исследовании, проведенным в Германии, приняли участие 1333 человека без нарушения функции ЩЖ. У лиц с уровнем ТТГ от 2,5 до 4,5 мЕд/л риск метаболического синдрома был повышен в 1,7 раза по сравнению с группой, где ТТГ был ближе к нижней границе нормы. Кроме того, приняв во внимание факт, что снижение функции ЩЖ может увеличивать риск ССЗ у пациентов с метаболическим синдромом, несколько проведенных исследований среди пациентов с нормальной функцией ЩЖ показали связь с традиционными и нетрадиционными факторами риска ССЗ. В популяционном исследовании в Нидерландах у лиц без нарушения функции ЩЖ уровень свободного Т4 обратно пропорционально коррелировал с триглицеридами, холестерином, АД, абдоминальным ожирением, инсулинорезистентностью. Увеличение индекса массы тела и уровня АД с увеличением уровня ТТГ было продемонстрировано в двух крупных популяционных исследованиях в Европе. Что касается дислипидемии, то у лиц без нарушения функции ЩЖ как с СД, так и без СД связь между инсулинорезистентностью и высоконормальным ТТГ определяла худший липидный профиль [2].
F. Amati с соавторами предоставили косвенные доказательства того, что у пациентов с СД и субклиническим гипотиреозом при отсутствии лечения последнего показатели сердечно-сосудистого риска хуже, чем в ситуации, когда гипотиреоз компенсировался. Для женщин с субклиническим гипотиреозом и женщин с нормальным уровнем тиреоидных гормонов с НТГ была разработана 16-недельная программа умеренных аэробных упражнений в сочетании с диетой. Важным результатом этого исследования стал тот факт, что благодаря разработанной программе восстановление чувствительности к инсулину и снижение массы тела произошло только в группе эутиреоза. Существуют данные о восстановлении чувствительности к инсулину при назначении терапии левотироксином пациентам с субклиническим гипотиреозом [18].
У пациентов с гипотиреозом и инсулинорезистентностью необходимо принимать во внимание влияние принимаемых ими препаратов. Так, метформин значимо снижает уровень ТТГ у пациентов с СД и гипотиреозом. Указанный эффект наблюдался в группе пациентов как с СД и гипотиреозом, получавших терапию левотироксином, так и в группе без терапии левотироксином, и у женщин с гипотиреозом и поликистозом яичников. Механизм такого влияния до сих пор неизвестен.
Инсулинорезистентность и структурные изменения щитовидной железы
Известно, что инсулин и инсулиноподобный фактор роста 1 могут стимулировать канцерогенез. Это частично объясняет причину, почему СД признан независимым фактором риска онкологических заболеваний. Вероятно, высокодифференцированный рак ЩЖ не является исключением из этого правила. Недавно полученные сообщения подтверждают, что пациенты с инсулинорезистентностью более склонны к развитию узлового зоба [19]. Более того, у пациентов с высокодифференцированным раком ЩЖ инсулинорезистентность встречается чаще [20]. Таким образом, необходимо помнить о влиянии высокого уровня ТТГ и гиперинсулинемии на пролиферацию тиреоцитов у пациентов с метаболическим синдромом.
Заключение
Гормоны ЩЖ оказывают влияние на углеводный обмен. Многим пациентам с СД приходится корректировать потребность в инсулине при развитии тиреотоксикоза или гипотиреоза. Своевременная коррекция нарушений функции ЩЖ приводит к восстановлению метаболических изменений. Одним из направлений в изучении СД является изучение возможной связи субклинических нарушений функции ЩЖ с инсулинорезистентностью.
Это связано с тем, что тиреоидные гормоны не только антагонисты инсулина, не только контринсулярные гормоны, но по влиянию на периферические ткани они являются синергистами инсулина и способствуют транспорту и утилизации глюкозы. Влияние тиреоидных гормонов на углеводный обмен зависит от преморбидного статуса, наследуемой предрасположенности к инсулинорезистентности и других факторов.
1. The 6th edition of the IDF Diabetes Atlas, last updated 2013.
2. Brenta G. Diabetes and thyroid disorders // British Journal of Diabetes & Vascular Disease. — 2010. — № 10(4). — Р. 172–177.
3. Kahn R., Buse J., Ferrannini E., Stern M.; American Diabetes Association; European Association for the Study of Diabetes. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes // Diabetes Care. — 2005. — № 28(9). — Р. 2289–304.
4. Cappola A.R., Ladenson P.W. Hypothyroidism and atherosclerosis // J. Clin. Endocrinol. Metab. — 2003. — № 88(6). — Р. 2438–44.
5. Biondi B., Cooper D.S. The clinical significance of subclinical thyroid dysfunction // Endocr. Rev. — 2008. — № 29(1). — Р. 76–131.
6. Паньків В.І., Гаврилюк В.М., Непорадна Л.Д. та ін. Взаємозв’язки між структурно-функціональними порушеннями щитоподібної залози і компонентами метаболічного синдрому // Міжнародний ендокринологічний журнал. — 2011. — № 6(38). — С. 39–43.
7. Beylot M., Laville M. Thyroid hormones and intermediary metabolism // The thyroid and tissues. — Schattauer, Stuttgart and New York, 1994. — Р. 47–59.
8. Duntas L.H., Orgiazzi J., Brabant G. The Interface between thyroid and diabetes mellitus // Clin. Endocrinol. (Oxf.). — 2011 Feb 24. — Doi 10.1111/j.1365–2265.2011.04029.
9. Weinberg M.B., Utter M.F. Effect of thyroid hormone on the turnover of rat liver pyruvate carboxylase and pyruvate dehydrogenase // J. Biol. Chem. — 1979. — № 254(19). — Р. 9492–9499.
10. Park E.A., Jerden D.C., Bahouth S.W. Regulation of phosphoenolpyruvate carboxykinase gene transcription by thyroid hormone involves two distinct binding sites in the promoter // Biochem. J. — 1995. — № 309(Pt. 3). — Р. 913–9.
11. Feng X., Jiang Y., Meltzer P., Yen P.M. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray // Mol. Endocrinol. — 2000. — № 14(7). — Р. 947–55.
12. Weinstein S.P., O’Boyle E., Fisher M., Haber R.S. Regulation of GLUT2 glucose transporter expression in liver by thyroid hormone: evidence for hormonal regulation of the hepatic glucose transport system // Endocrinology. — 1994. — № 135(2). — Р. 649–54.
13. Klieverik L.P., Janssen S.F., van Riel A. et al. Thyroid hormone modulates glucose production via a sympathetic pathway from the hypothalamic paraventricular nucleus to the liver // Proc. Natl. Acad. Sci. U. S. A. — 2009. — № 106. — Р. 5966–5971.
14. Brenta G. Diabetes and thyroid disorders // Thyroid. International. — 2011. — № 3.
15. Handisurya A., Pacini G., Tura A. et al. Effects of thyroxine replacement therapy on glucose metabolism in subjects with subclinical and overt hypothyroidism // Clin. Endocrinol. (Oxf.). — 2008. — № 69(6). — Р. 963–9.
16. Okosieme O., Wijeyaratne C., Lazarus J., Premawardhana L. Restricted thyroglobulin antibody epitope specificities in subjects with type 1 diabetes mellitus // Eur. J. Endocrinol. — 2009. — № 161(3). — Р. 489–93.
17. Mohn A., Di Michele S., Di Luzio R. et al. The effect of subclinical hypothyroidism on metabolic control in children and adolescents with Type 1 diabetes mellitus // Diabet Med. — 2002. — № 19(1). — Р. 70–3.
18. Amati F., Dubе J.J., Stefanovic-Racic M. et al. Improvements in insulin sensitivity are blunted by subclinical hypothyroidism // Med. Sci. Sports Exerc. — 2009. — № 41(2). — Р. 265–9.
19. Rezzónico J., Rezzónico M., Pusiol E. et al. Introducing the thyroid gland as another victim of the insulin resistance syndrome // Thyroid. — 2008. — № 18(4). — Р. 461–4.
20. Rezzónico J.N., Rezzónico M., Pusiol E. et al. Increased prevalence of insulin resistance in patients with differentiated thyroid carcinoma // Metab. Syndr. Relat. Disord. — 2009. — № 7(4). — Р. 375–80.